Population Structure of Lassa Mammarenavirus in West Africa



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
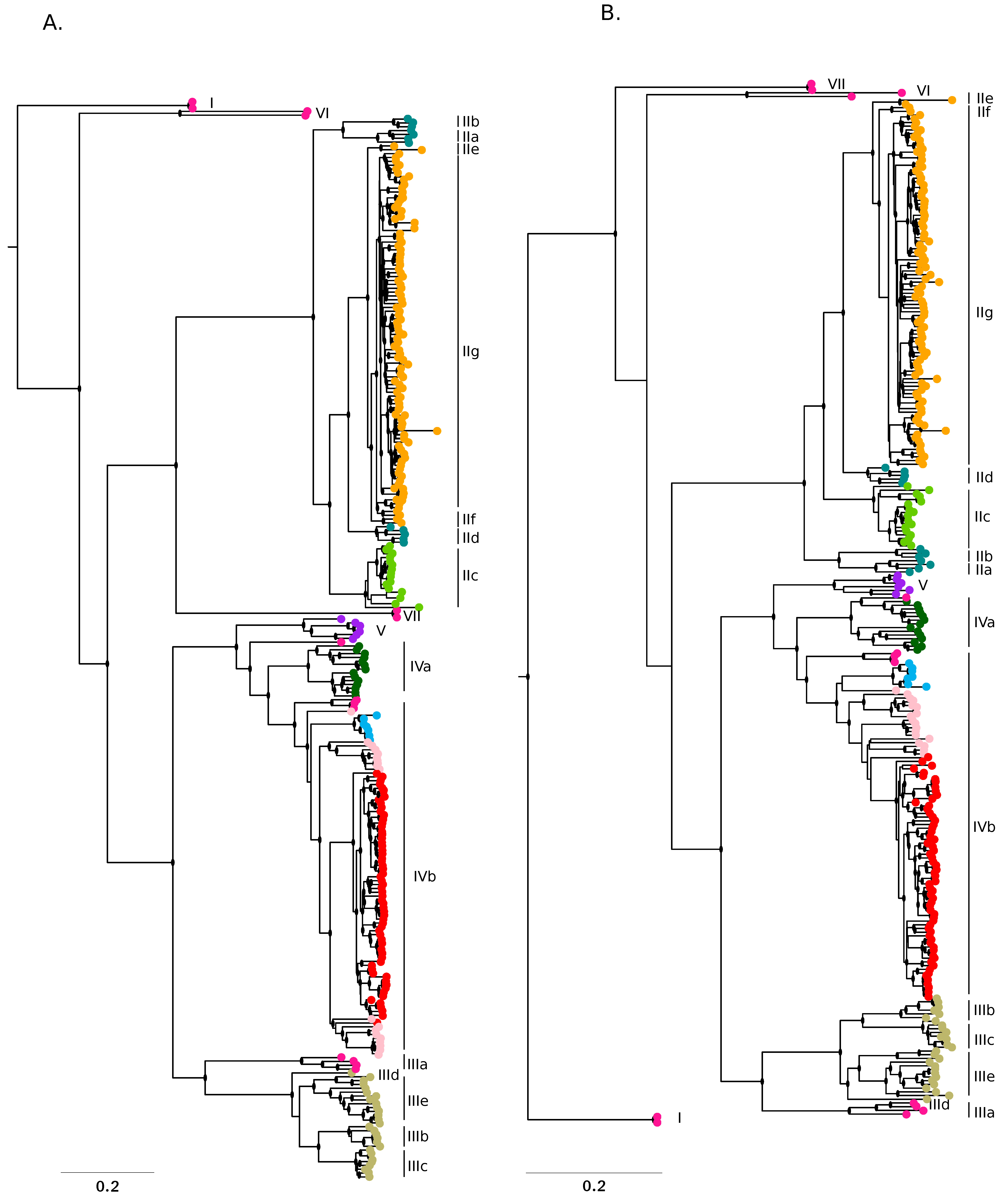
2.1. Sequences, Alignments, and Phylogenetic Trees
2.2. Recombination
2.3. Linkage Disequilibrium and Population Structure
2.4. DAPC Analysis
2.5. Statistical Analysis of Disease Outcome and Viral Loads
3. Results
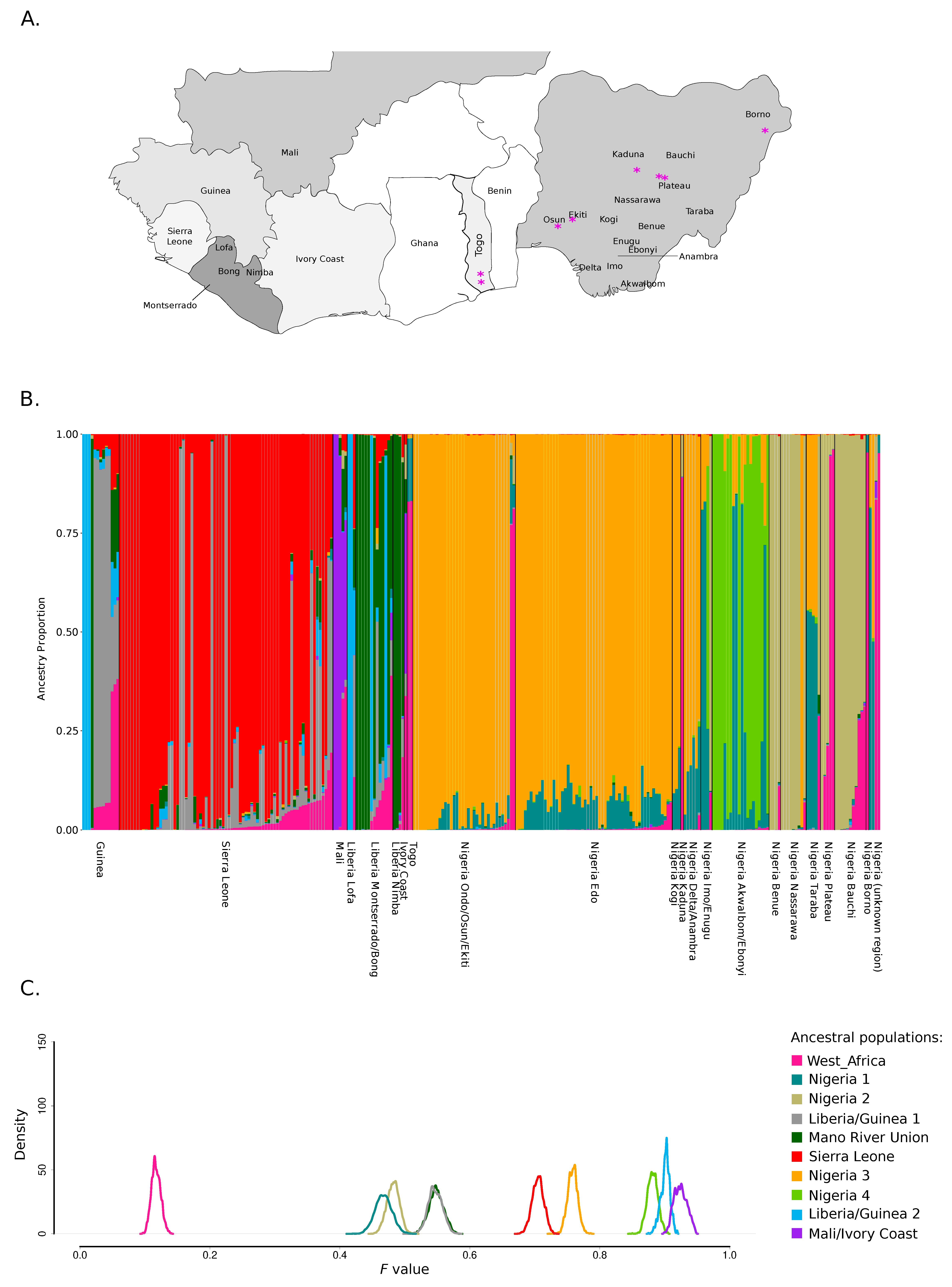
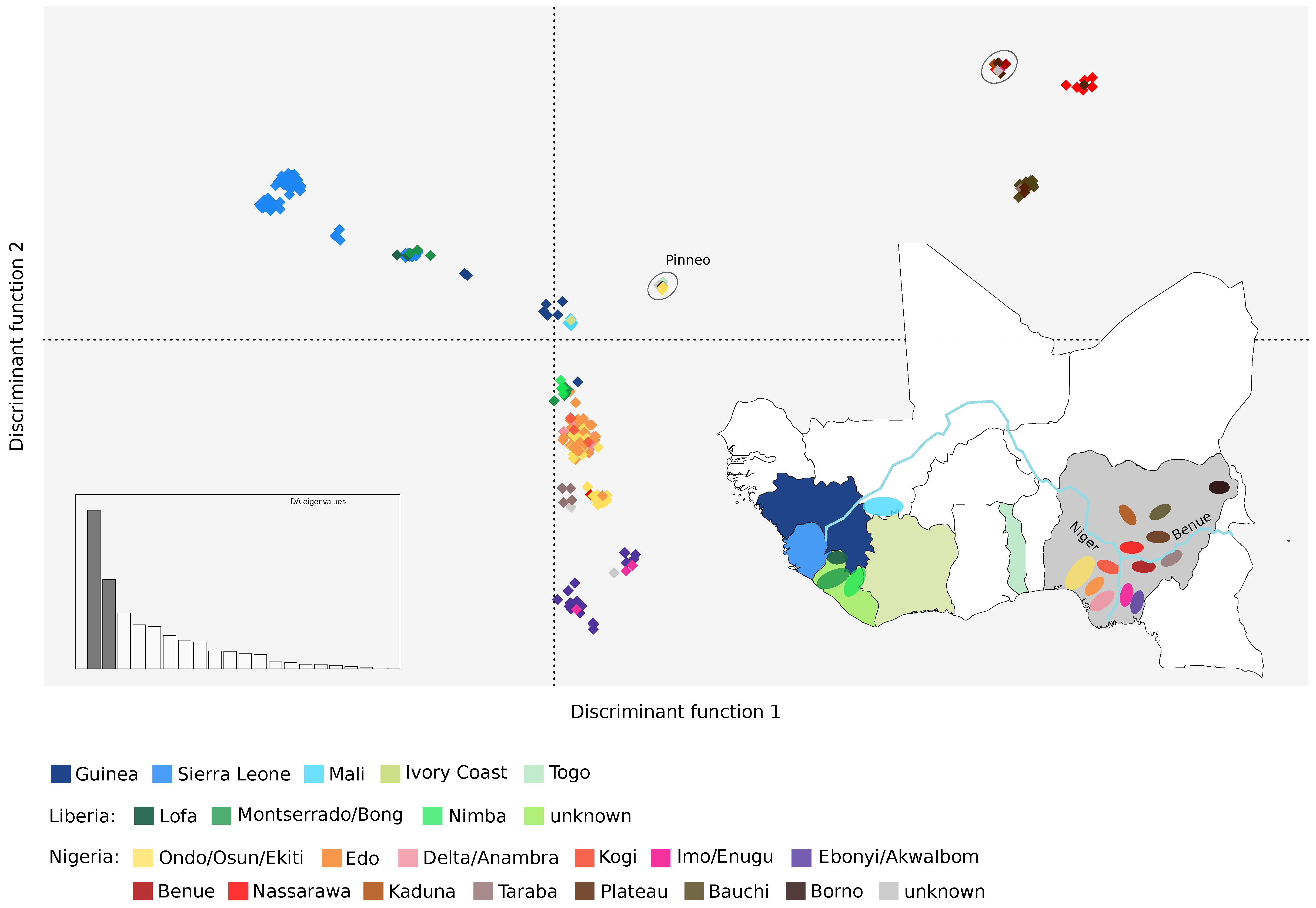
3.1. Population Structure of LASV in West Africa
3.2. Geographic Gradients of LASV Diversity
3.3. Population Structure of LASV Lineage II and Association with Disease Severity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Happi, A.N.; Happi, C.T.; Schoepp, R.J. Lassa Fever Diagnostics: Past, Present, and Future. Curr. Opin. Virol. 2019, 37, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, S.L.M.; Strecker, T.; Cadar, D.; Dienes, H.P.; Faber, K.; Patel, K.; Brown, S.M.; Davis, W.G.; Klena, J.D.; Rollin, P.E.; et al. New Lineage of Lassa Virus, Togo, 2016. Emerg. Infect. Dis. 2018, 24, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Salu, O.B.; James, A.B.; Bankole, H.S.; Agbla, J.M.; Da Silva, M.; Gbaguidi, F.; Loko, C.F.; Omilabu, S.A. Molecular Confirmation and Phylogeny of Lassa Fever Virus in Benin Republic 2014–2016. Afr. J. Lab. Med. 2019, 8, 803. [Google Scholar] [CrossRef]
- Frame, J.D.; Baldwin, J.M., Jr.; Gocke, D.J.; Troup, J.M. Lassa Fever, a New Virus Disease of Man from West Africa. I. Clinical Description and Pathological Findings. Am. J. Trop. Med. Hyg. 1970, 19, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Charrel, R.N.; de Lamballerie, X. Zoonotic Aspects of Arenavirus Infections. Vet. Microbiol. 2010, 140, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pontremoli, C.; Forni, D.; Sironi, M. Arenavirus Genomics: Novel Insights into Viral Diversity, Origin, and Evolution. Curr. Opin. Virol. 2019, 34, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Olayemi, A.; Cadar, D.; Magassouba, N.; Obadare, A.; Kourouma, F.; Oyeyiola, A.; Fasogbon, S.; Igbokwe, J.; Rieger, T.; Bockholt, S.; et al. New Hosts of the Lassa Virus. Sci. Rep. 2016, 6, 25280. [Google Scholar] [CrossRef]
- Yadouleton, A.; Agolinou, A.; Kourouma, F.; Saizonou, R.; Pahlmann, M.; Bedie, S.K.; Bankole, H.; Becker-Ziaja, B.; Gbaguidi, F.; Thielebein, A.; et al. Lassa Virus in Pygmy Mice, Benin, 2016–2017. Emerg. Infect. Dis. 2019, 25, 1977–1979. [Google Scholar] [CrossRef]
- Colangelo, P.; Verheyen, E.; Leirs, H.; Tatard, C.; Denys, C.; Dobigny, G.; Duplantier, J.M.; Brouat, C.; Granjon, L.; Lecompte, E. A Mitochondrial Phylogeographic Scenario for the most Widespread African Rodent, Mastomys Natalensis. Biol. J. Linn. Soc. Lond. 2013, 108, 901–916. [Google Scholar] [CrossRef]
- Fichet-Calvet, E.; Audenaert, L.; Barrière, P.; Verheyen, E. Diversity, Dynamics and Reproduction in a Community of Small Mammals in Upper Guinea, with Emphasis on Pygmy Mice Ecology. Afr. J. Ecol. 2009, 48, 600–614. [Google Scholar] [CrossRef]
- Bonwitt, J.; Saez, A.M.; Lamin, J.; Ansumana, R.; Dawson, M.; Buanie, J.; Lamin, J.; Sondufu, D.; Borchert, M.; Sahr, F.; et al. At Home with Mastomys and Rattus: Human-Rodent Interactions and Potential for Primary Transmission of Lassa Virus in Domestic Spaces. Am. J. Trop. Med. Hyg. 2017, 96, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Brouat, C.; Loiseau, A.; Kane, M.; Ba, K.; Duplantier, J.M. Population Genetic Structure of Two Ecologically Distinct Multimammate Rats: The Commensal Mastomys Natalensis and the Wild Mastomys Erythroleucus in Southeastern Senegal. Mol. Ecol. 2007, 16, 2985–2997. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.R.; Sole, C.L.; Barbato, M.; von Bramann, U.; Bruford, M.W. Landscape Determinants of Fine-Scale Genetic Structure of a Small Rodent in a Heterogeneous Landscape (Hluhluwe-iMfolozi Park, South Africa). Sci. Rep. 2016, 6, 29168. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Rollin, P.E.; Ksiazek, T.G.; Hustad, H.L.; Bausch, D.G.; Demby, A.H.; Bajani, M.D.; Peters, C.J.; Nichol, S.T. Genetic Diversity among Lassa Virus Strains. J. Virol. 2000, 74, 6992–7004. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Shapiro, B.J.; Matranga, C.B.; Sealfon, R.; Lin, A.E.; Moses, L.M.; Folarin, O.A.; Goba, A.; Odia, I.; Ehiane, P.E.; et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 2015, 162, 738–750. [Google Scholar] [CrossRef] [PubMed]
- Ehichioya, D.U.; Hass, M.; Becker-Ziaja, B.; Ehimuan, J.; Asogun, D.A.; Fichet-Calvet, E.; Kleinsteuber, K.; Lelke, M.; ter Meulen, J.; Akpede, G.O.; et al. Current Molecular Epidemiology of Lassa Virus in Nigeria. J. Clin. Microbiol. 2011, 49, 1157–1161. [Google Scholar] [CrossRef]
- Siddle, K.J.; Eromon, P.; Barnes, K.G.; Mehta, S.; Oguzie, J.U.; Odia, I.; Schaffner, S.F.; Winnicki, S.M.; Shah, R.R.; Qu, J.; et al. Genomic Analysis of Lassa Virus during an Increase in Cases in Nigeria in 2018. N. Engl. J. Med. 2018, 379, 1745–1753. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Pullan, S.T.; Lemey, P.; Suchard, M.A.; Ehichioya, D.U.; Pahlmann, M.; Thielebein, A.; Hinzmann, J.; Oestereich, L.; Wozniak, D.M.; et al. Metagenomic Sequencing at the Epicenter of the Nigeria 2018 Lassa Fever Outbreak. Science 2019, 363, 74–77. [Google Scholar] [CrossRef]
- Ehichioya, D.U.; Dellicour, S.; Pahlmann, M.; Rieger, T.; Oestereich, L.; Becker-Ziaja, B.; Cadar, D.; Ighodalo, Y.; Olokor, T.; Omomoh, E.; et al. Phylogeography of Lassa Virus in Nigeria. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Wiley, M.R.; Fakoli, L.; Letizia, A.G.; Welch, S.R.; Ladner, J.T.; Prieto, K.; Reyes, D.; Espy, N.; Chitty, J.A.; Pratt, C.B.; et al. Lassa Virus Circulating in Liberia: A Retrospective Genomic Characterisation. Lancet Infect. Dis. 2019, 19, 1371–1378. [Google Scholar] [CrossRef]
- Manning, J.T.; Forrester, N.; Paessler, S. Lassa Virus Isolates from Mali and the Ivory Coast Represent an Emerging Fifth Lineage. Front. Microbiol. 2015, 6, 1037. [Google Scholar] [CrossRef] [PubMed]
- Pontremoli, C.; Forni, D.; Cagliani, R.; Pozzoli, U.; Riva, S.; Bravo, I.G.; Clerici, M.; Sironi, M. Evolutionary Analysis of Old World Arenaviruses Reveals a Major Adaptive Contribution of the Viral Polymerase. Mol. Ecol. 2017, 26, 5173–5188. [Google Scholar] [CrossRef] [PubMed]
- Wauquier, N.; Petitdemange, C.; Tarantino, N.; Maucourant, C.; Coomber, M.; Lungay, V.; Bangura, J.; Debre, P.; Vieillard, V. HLA-C-Restricted Viral Epitopes are Associated with an Escape Mechanism from KIR2DL2(+) NK Cells in Lassa Virus Infection. EBioMedicine 2019, 40, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Okokhere, P.; Colubri, A.; Azubike, C.; Iruolagbe, C.; Osazuwa, O.; Tabrizi, S.; Chin, E.; Asad, S.; Ediale, E.; Rafiu, M.; et al. Clinical and Laboratory Predictors of Lassa Fever Outcome in a Dedicated Treatment Facility in Nigeria: A Retrospective, Observational Cohort Study. Lancet Infect. Dis. 2018, 18, 684–695. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum Likelihood-Based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar]
- Sawyer, S. Statistical Tests for Detecting Gene Conversion. Mol. Biol. Evol. 1989, 6, 526–538. [Google Scholar]
- Martin, D.; Rybicki, E. RDP: Detection of Recombination Amongst Aligned Sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the Mosaic Structure of Genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Bay, R.A.; Bielawski, J.P. Recombination Detection Under Evolutionary Scenarios Relevant to Functional Divergence. J. Mol. Evol. 2011, 73, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Haubold, B.; Hudson, R.R. LIAN 3.0: Detecting Linkage Disequilibrium in Multilocus Data. Linkage Analysis. Bioinformatics 2000, 16, 847–848. [Google Scholar] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Wang, J. The Computer Program Structure for Assigning Individuals to Populations: Easy to use but Easier to Misuse. Mol. Ecol. Resour. 2017, 17, 981–990. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals using the Software STRUCTURE: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Janes, J.K.; Miller, J.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Cullingham, C.I.; Andrew, R.L. The K = 2 Conundrum. Mol. Ecol. 2017, 26, 3594–3602. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A Website and Program for Visualizing STRUCTURE Output and Implementing the Evanno Method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A Program for Identifying Clustering Modes and Packaging Population Structure Inferences Across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant Analysis of Principal Components: A New Method for the Analysis of Genetically Structured Populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: http://www.r-project.org (accessed on 20 February 2020).
- Puechmaille, S.J. The Program Structure does Not Reliably Recover the Correct Population Structure when Sampling is Uneven: Subsampling and New Estimators Alleviate the Problem. Mol. Ecol. Resour. 2016, 16, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Grande-Perez, A.; Martin, V.; Moreno, H.; de la Torre, J.C. Arenavirus Quasispecies and their Biological Implications. Curr. Top. Microbiol. Immunol. 2016, 392, 231–276. [Google Scholar] [PubMed]
- Szmaragd, C.; Balloux, F. The Population Genomics of Hepatitis B Virus. Mol. Ecol. 2007, 16, 4747–4758. [Google Scholar] [CrossRef] [PubMed]
- Olayemi, A.; Obadare, A.; Oyeyiola, A.; Igbokwe, J.; Fasogbon, A.; Igbahenah, F.; Ortsega, D.; Asogun, D.; Umeh, P.; Vakkai, I.; et al. Arenavirus Diversity and Phylogeography of Mastomys Natalensis Rodents, Nigeria. Emerg. Infect. Dis. 2016, 22, 694–697. [Google Scholar] [CrossRef]
- Nikisins, S.; Rieger, T.; Patel, P.; Muller, R.; Gunther, S.; Niedrig, M. International External Quality Assessment Study for Molecular Detection of Lassa Virus. PLoS Negl. Trop. Dis. 2015, 9, e0003793. [Google Scholar] [CrossRef]
- Asogun, D.A.; Adomeh, D.I.; Ehimuan, J.; Odia, I.; Hass, M.; Gabriel, M.; Olschlager, S.; Becker-Ziaja, B.; Folarin, O.; Phelan, E.; et al. Molecular Diagnostics for Lassa Fever at Irrua Specialist Teaching Hospital, Nigeria: Lessons Learnt from Two Years of Laboratory Operation. PLoS Negl. Trop. Dis. 2012, 6, e1839. [Google Scholar] [CrossRef]
- Gryseels, S.; Baird, S.J.; Borremans, B.; Makundi, R.; Leirs, H.; de Gouy Bellocq, J. When Viruses Don’t Go Viral: The Importance of Host Phylogeographic Structure in the Spatial Spread of Arenaviruses. PLoS Pathog. 2017, 13, e1006073. [Google Scholar] [CrossRef]
- Kronmann, K.C.; Nimo-Paintsil, S.; Guirguis, F.; Kronmann, L.C.; Bonney, K.; Obiri-Danso, K.; Ampofo, W.; Fichet-Calvet, E. Two Novel Arenaviruses Detected in Pygmy Mice, Ghana. Emerg. Infect. Dis. 2013, 19, 1832–1835. [Google Scholar] [CrossRef]
- McLay, L.; Lan, S.; Ansari, A.; Liang, Y.; Ly, H. Identification of Virulence Determinants within the L Genomic Segment of the Pichinde Arenavirus. J. Virol. 2013, 87, 6635–6643. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Emonet, S.F.; Welch, M.J.; Lee, A.M.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Point Mutation in the Glycoprotein of Lymphocytic Choriomeningitis Virus is Necessary for Receptor Binding, Dendritic Cell Infection, and Long-Term Persistence. Proc. Natl. Acad. Sci. USA 2011, 108, 2969–2974. [Google Scholar] [CrossRef]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de La Torre, J.C.; Oldstone, M.B. Immunosuppression and Resultant Viral Persistence by Specific Viral Targeting of Dendritic Cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Riviere, Y.; Ahmed, R.; Southern, P.J.; Buchmeier, M.J.; Oldstone, M.B. Genetic Mapping of Lymphocytic Choriomeningitis Virus Pathogenicity: Virulence in Guinea Pigs is Associated with the L RNA Segment. J. Virol. 1985, 55, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Djavani, M.; Topisirovic, I.; Zapata, J.C.; Sadowska, M.; Yang, Y.; Rodas, J.; Lukashevich, I.S.; Bogue, C.W.; Pauza, C.D.; Borden, K.L.; et al. The Proline-Rich Homeodomain (PRH/HEX) Protein is Down-Regulated in Liver during Infection with Lymphocytic Choriomeningitis Virus. J. Virol. 2005, 79, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Lohning, M.; Pinschewer, D.D. Viral Replicative Capacity is the Primary Determinant of Lymphocytic Choriomeningitis Virus Persistence and Immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 21641–21646. [Google Scholar] [CrossRef]
- Bergthaler, A.; Merkler, D.; Horvath, E.; Bestmann, L.; Pinschewer, D.D. Contributions of the Lymphocytic Choriomeningitis Virus Glycoprotein and Polymerase to Strain-Specific Differences in Murine Liver Pathogenicity. J. Gen. Virol. 2007, 88, 592–603. [Google Scholar] [CrossRef]
- Ng, C.T.; Sullivan, B.M.; Oldstone, M.B. The Role of Dendritic Cells in Viral Persistence. Curr. Opin. Virol. 2011, 1, 160–166. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forni, D.; Sironi, M. Population Structure of Lassa Mammarenavirus in West Africa. Viruses 2020, 12, 437. https://doi.org/10.3390/v12040437
Forni D, Sironi M. Population Structure of Lassa Mammarenavirus in West Africa. Viruses. 2020; 12(4):437. https://doi.org/10.3390/v12040437
Chicago/Turabian StyleForni, Diego, and Manuela Sironi. 2020. "Population Structure of Lassa Mammarenavirus in West Africa" Viruses 12, no. 4: 437. https://doi.org/10.3390/v12040437
APA StyleForni, D., & Sironi, M. (2020). Population Structure of Lassa Mammarenavirus in West Africa. Viruses, 12(4), 437. https://doi.org/10.3390/v12040437