The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid



Abstract
1. Introduction
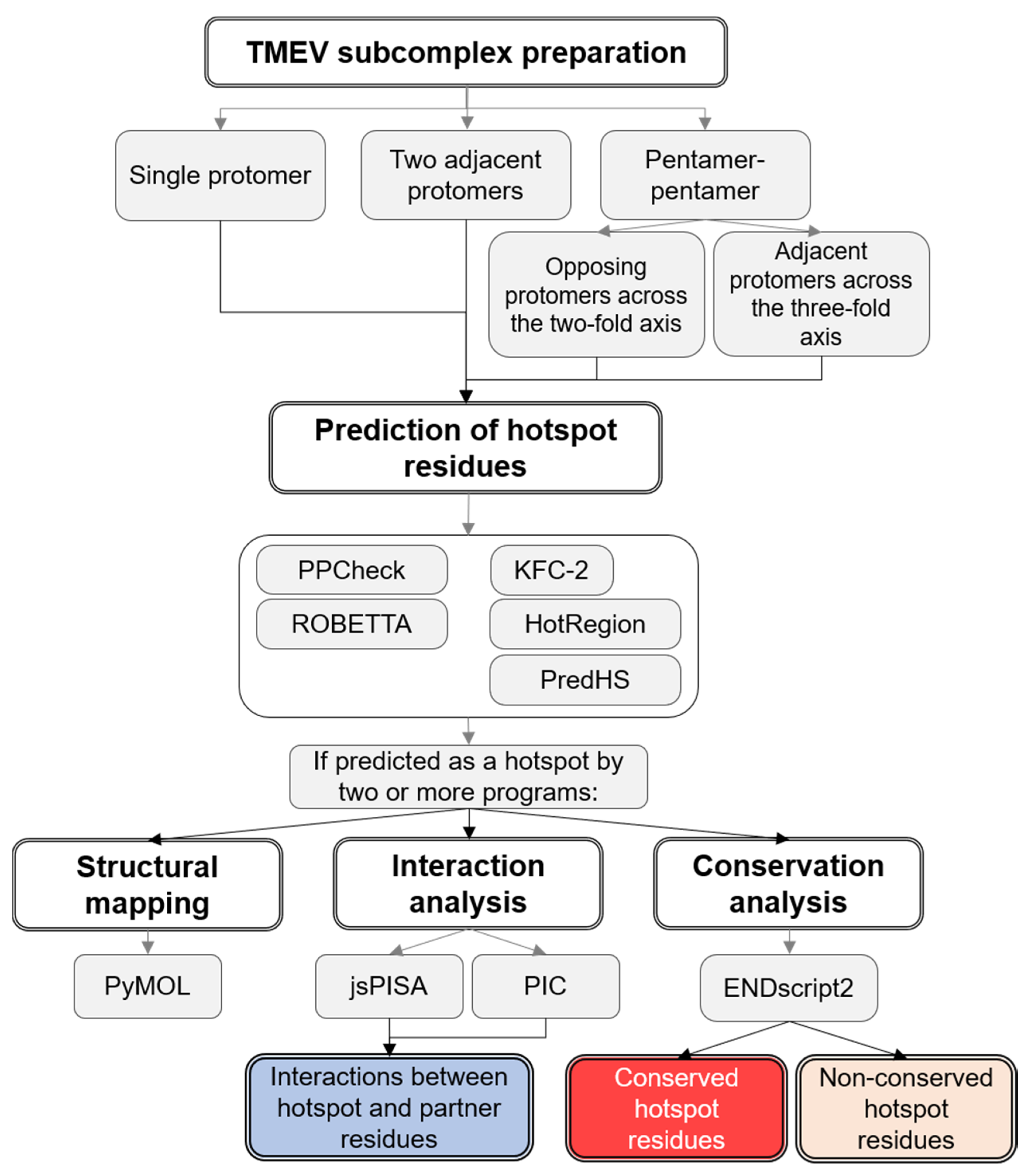
2. Materials and Methods
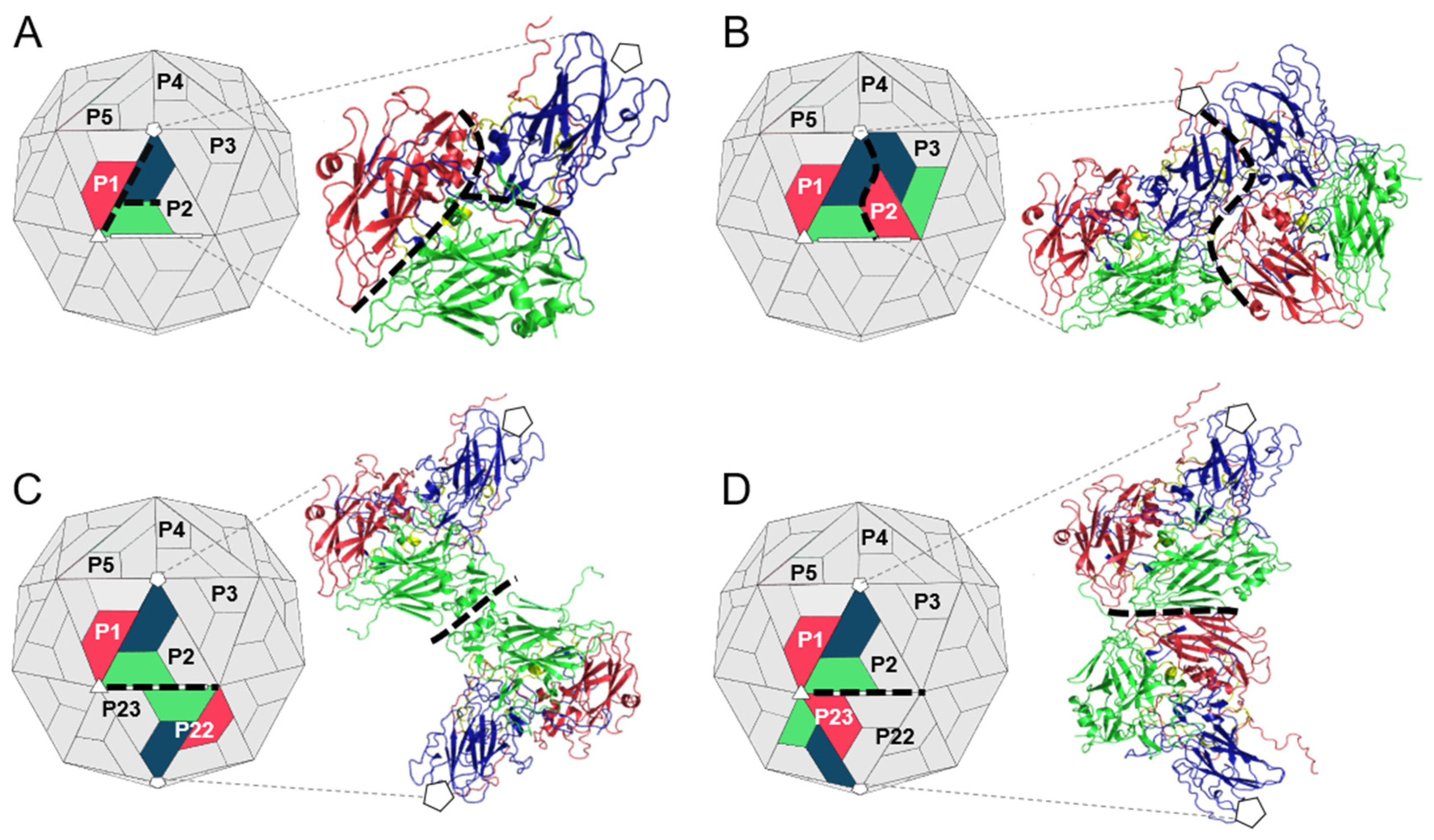
2.1. Preparation of TMEV Subcomplexes
2.2. Hotspot Prediction
2.3. Prediction of Interacting Residues
2.4. Residue Conservation
3. Results
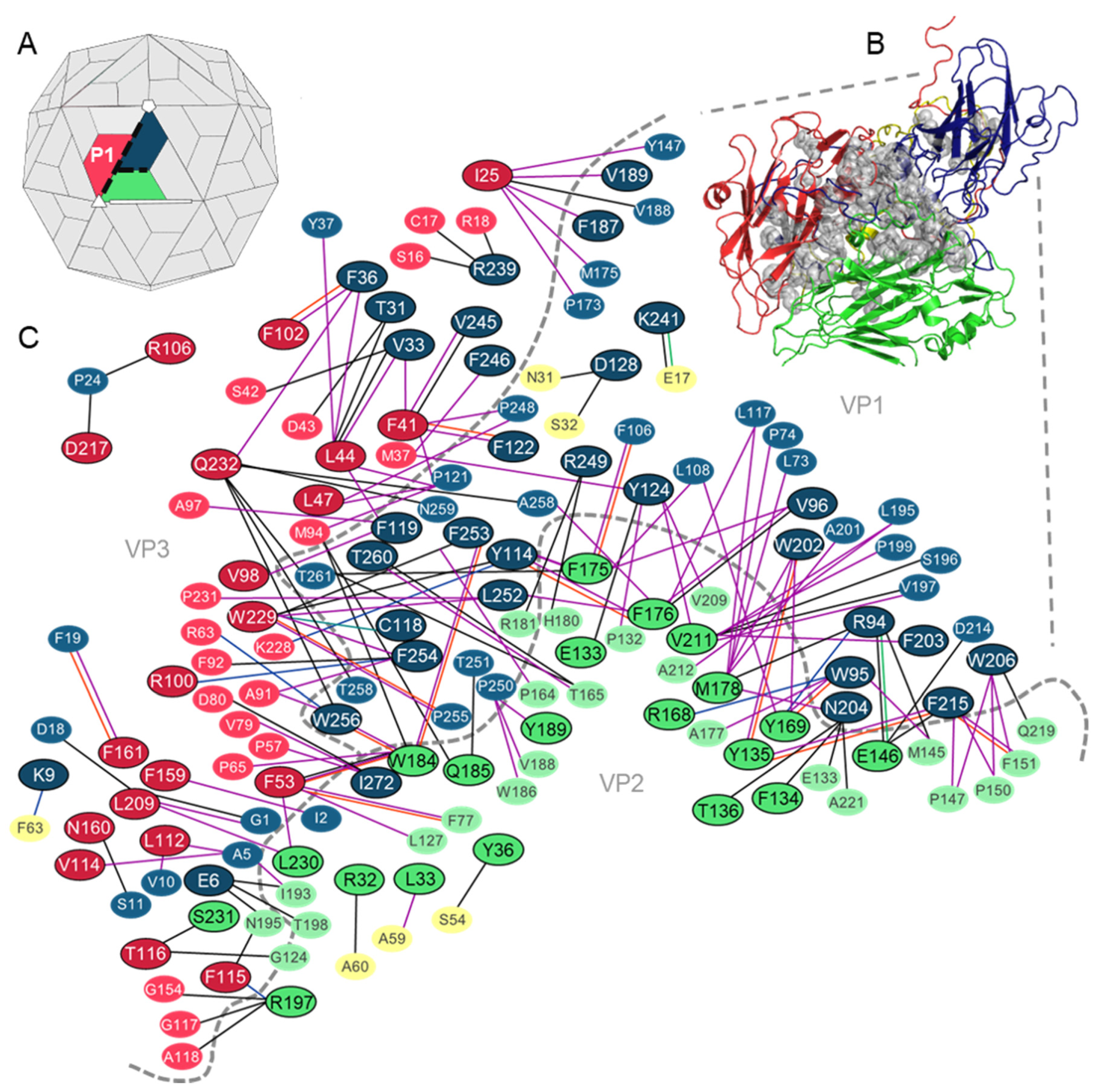
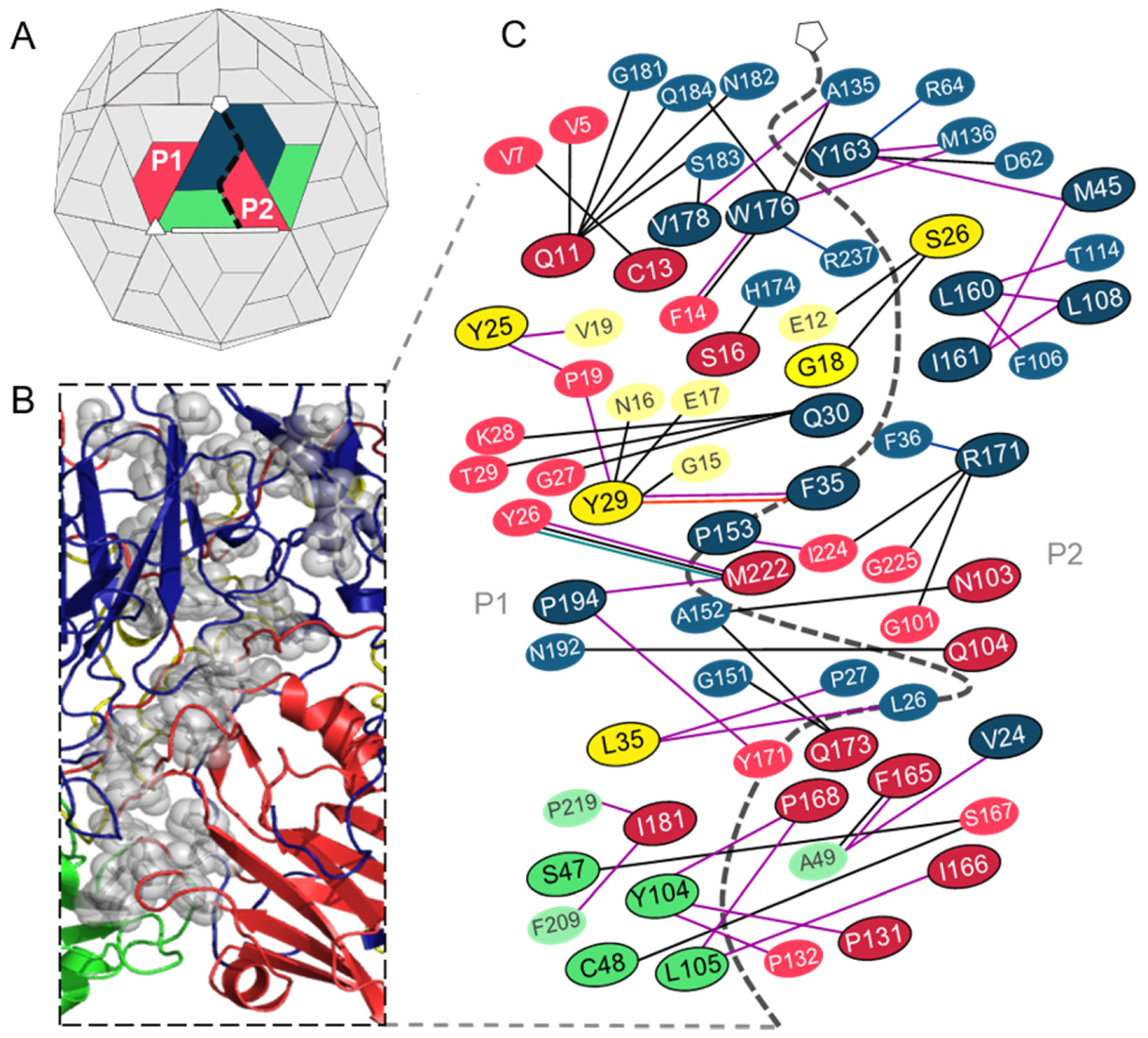
3.1. Analysis of the Residues that Contribute to the Intraprotomer Interfaces of TMEV GDVII
3.1.1. Predicted Hotspots at the Intraprotomer Interfaces
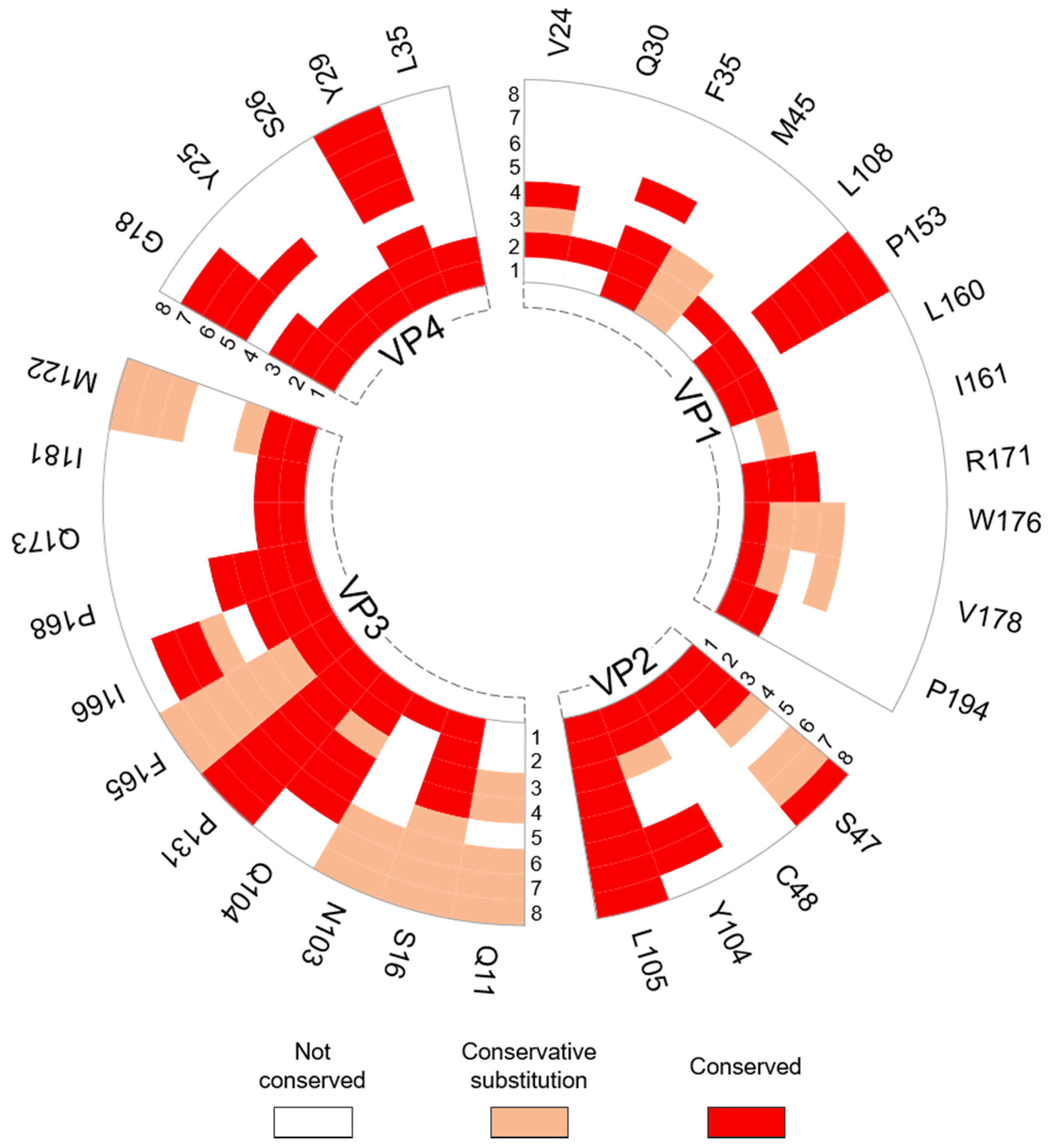
3.1.2. Intraprotomer Hotspot Residues in TMEV that are Conserved with Residues in Other Viruses of the Family
3.2. Analysis of the Residues that Contribute to the Interprotomer Interfaces of TMEV GDVII
3.2.1. Predicted Hotspot Residues at the Interprotomer Interfaces
3.2.2. Interprotomer Hotspot Residues in TMEV that are Conserved with Residues in Other Viruses of the Family
3.3. Analysis of the Residues that Contribute to the Interpentamer Interfaces of TMEV GDVII
3.3.1. Predicted Hotspot Residues at the Interpentamer Interfaces
3.3.2. Interpentamer Hotspot Residues in TMEV that are Conserved with Residues in Other Viruses of the Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zell, R. Picornaviridae - the ever-growing virus family. Arch. Virol. 2017, 163, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, A.L.; Baggs, E.L.; Semler, B.L. Picornaviruses: Pathogenesis and Molecular Biology. In Reference Module in Biomedical Research; Caplan, M., Ed.; Elsevier: Irvine, CA, USA, 2015; pp. 1–11. ISBN 9780128012383. [Google Scholar]
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-Spectrum Antivirals against 3C or 3C-Like Proteases of Picornaviruses, Noroviruses, and Coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, K.C.; Susi, P.; Jochmans, D.; Koskinen, J.; Landt, O.; Sanchez, N.; Palm, K.; Neyts, J.; Butcher, S.J. Progress in human picornavirus research: New findings from the AIROPico consortium. Antiviral Res. 2019, 161, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Cifuente, J.O.; Moratorio, G. Evolutionary and Structural Overview of Human Picornavirus Capsid Antibody Evasion. Front. Cell. Infect. Microbiol. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; Ott, J.J.; Van Damme, P.; Shouval, D. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 2018, 68, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J.; Kristensen, T.; Jackson, T. Foot-and-mouth disease virus: Prospects for using knowledge of virus biology to improve control of this continuing global threat. Virus Res. 2020, 281, 197909. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Liu, Y.; Ma, H.C.; Paul, A.; Wimmer, E. Picornavirus Morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef]
- Rincón, V.; Rodríguez-Huete, A.; Mateu, M.G. Different functional sensitivity to mutation at intersubunit interfaces involved in consecutive stages of foot-and-mouth disease virus assembly. J. Gen. Virol. 2015, 96, 2595–2606. [Google Scholar] [CrossRef]
- Mateu, M.G. Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 2013, 531, 65–79. [Google Scholar] [CrossRef]
- Mateo, R.; Diaz, A.; Baranowski, E.; Mateu, M.G. Complete Alanine Scanning of Intersubunit Interfaces in a Foot-and-Mouth Disease Virus Capsid Reveals Critical Contributions of Many Side Chains to Particle Stability and Viral Function. J. Biol. Chem. 2003, 278, 41019–41027. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J. A hot spot of binding energy in a horomone-receptor interface. Science (80-.) 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot spots—A review of the protein – protein interface determinant amino-acid residues. Proteins Struct. Funct. Bioinforma. 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Mateu, M.G. The capsid protein of human immunodeficiency virus: Intersubunit interactions during virus assembly. FEBS J. 2009, 276, 6098–6109. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.M.; Rambhia, P.H.; French, R.H.; Steinmetz, N.F. Design rules for nanomedical engineering: From physical virology to the applications of virus-based materials in medicine. J. Biol. Phys. 2013, 39, 301–325. [Google Scholar] [CrossRef] [PubMed]
- Fox, H.; Knowlson, S.; Minor, P.D.; Macadam, A.J. Genetically Thermo-Stabilised, Immunogenic Poliovirus Empty Capsids; a Strategy for Non-replicating Vaccines. PLoS Pathog. 2017, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Warwicker, J. Model for the differential stabilities of rhinovirus and poliovirus to mild acidic pH, based on electrostatics calculations. J. Mol. Biol. 1992, 223, 247–257. [Google Scholar] [CrossRef]
- Maree, F.F.; Blignaut, B.; de Beer, T.A.P.; Rieder, E. Analysis of SAT Type Foot-And-Mouth Disease Virus Capsid Proteins and the Identification of Putative Amino Acid Residues Affecting Virus Stability. PLoS ONE 2013, 8, e61612. [Google Scholar] [CrossRef]
- Caridi, F.; Vázquez-Calvo, A.; Sobrino, F.; Martín-Acebes, M.A. The pH Stability of Foot-and-Mouth Disease Virus Particles Is Modulated by Residues Located at the Pentameric Interface and in the N Terminus of VP1. J. Virol. 2015, 89, 5633–5642. [Google Scholar] [CrossRef]
- Yuan, S.; Li, G.; Wang, Y.; Gao, Q.; Wang, Y.; Cui, R.; Altmeyer, R.; Zou, G. Identification of Positively Charged Residues in Enterovirus 71 Capsid Protein VP1 Essential for Production of Infectious Particles. J. Virol. 2016, 90, 741–752. [Google Scholar] [CrossRef]
- Van Vlijmen, H.W.T.; Curry, S.; Schaefer, M.; Karplus, M. Titration Calculations of Foot-and-mouth Disease Virus Capsids and their Stabilities as a Function of pH. J. Mol. Biol. 1998, 275, 295–308. [Google Scholar] [CrossRef]
- Kotecha, A.; Seago, J.; Scott, K.; Burman, A.; Loureiro, S.; Ren, J.; Porta, C.; Ginn, H.M.; Jackson, T.; Perez-Martin, E.; et al. Structure-based energetics of protein interfaces guides foot-and-mouth disease virus vaccine design. Nat. Struct. Mol. Biol. 2015, 22, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Kortemme, T.; Kim, D.E.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 219, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Massova, I.; Kollman, P.A. Computational Alanine Scanning of the 1:1 Human Growth Hormone-Receptor Complex. J. Comput. Chem. 2002, 23, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wisniewski, J.A.; Ji, H. Hot spot-based design of small-molecule inhibitors for protein-protein interactions. Bioorg. Med. Chem. Lett. 2014, 24, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Rajamani, D.; Thiel, S.; Vajda, S.; Camacho, C.J. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2004, 101, 11287–11292. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Zhu, C.; Wang, L.; Abel, R.; Negron, C.; Pearlman, D.; Feyfant, E.; Duan, J.; Sherman, W. Predicting the Effect of Amino Acid Single-Point Mutations on Protein Stability - Large-Scale Validation of MD-Based Relative Free Energy Calculations. J. Mol. Biol. 2017, 429, 948–963. [Google Scholar] [CrossRef]
- Xia, J.; Zhao, X.; Song, J.; Huang, D. APIS: Accurate prediction of hot spots in protein interfaces by combining protrusion index with solvent accessibility. BMC Bioinform. 2010, 11, 1–14. [Google Scholar] [CrossRef]
- Liu, S.; Liu, C.; Deng, L. Machine learning approaches for protein-protein interaction hot spot prediction: Progress and comparative assessment. Molecules 2018, 23, 2535. [Google Scholar] [CrossRef]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot spots in protein-protein interfaces: Towards drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef]
- Qiao, Y.; Xiong, Y.; Gao, H.; Zhu, X.; Cheng, P. Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinform. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Zhang, S. Computational Prediction of Hot Spot Residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Kar, G.; Keskin, O.; Gursoy, A.; Nussinov, R. A survey of available tools and web servers for analysis of protein-protein interactions and interfaces. Brief. Bioinform. 2009, 10, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Gursoy, A.; Keskin, O. Identification of computational hot spots in protein interfaces: Combining solvent accessibility and inter-residue potentials improves the accuracy. Bioinformatics 2009, 25, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Knox, C.; Bishop Tastan, O. Interacting motif networks located in hotspots associated with RNA release are conserved in Enterovirus capsids. FEBS Lett. 2017, 591, 1687–1701. [Google Scholar] [CrossRef]
- Ross, C.; Upfold, N.; Luke, G.A.; Tastan, Ö.; Knox, C. Subcellular localisation of Theiler’s murine encephalomyelitis virus (TMEV) capsid subunit VP1 vis-á-vis host protein Hsp90. Virus Res. 2016, 222, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Upfold, N.; Ross, C.; Bishop, Ö.T.; Luke, G.A.; Knox, C. The generation and characterisation of neutralising antibodies against the Theiler’s murine encephalomyelitis virus (TMEV) GDVII capsid reveals the potential binding site of the host cell co-receptor, heparan sulfate. Virus Res. 2018, 244, 153–163. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 1.7.2.1; SchrÖdinger, LLC.: New York, NY, USA, 2010.
- Sukhwal, A.; Sowdhamini, R. PPCheck: A Webserver for the Quantitative Analysis of Protein–Protein Interfaces and Prediction of Residue Hotspots. Bioinform. Biol. Insights 2015, 9, 141–151. [Google Scholar] [CrossRef]
- Deng, L.; Zhang, Q.C.; Chen, Z.; Meng, Y.; Guan, J.; Zhou, S. PredHS: A web server for predicting protein-protein interaction hot spots by using structural neighborhood properties. Nucleic Acids Res. 2014, 1, 1–6. [Google Scholar] [CrossRef]
- Darnell, S.J.; Legault, L.; Mitchell, J.C. KFC Server: Interactive forecasting of protein interaction hot spots. Nucleic Acids Res. 2008, 36, W265–W269. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; Page, D.; Mitchell, J.C. An automated decision-tree approach to predicting protein interaction hot spots. Proteins 2007, 68, 813–823. [Google Scholar] [CrossRef]
- Cukuroglu, E.; Gursoy, A.; Keskin, O. HotRegion: A database of predicted hot spot clusters. Nucleic Acids Res. 2012, 40, D829–D833. [Google Scholar] [CrossRef] [PubMed]
- Tina, K.G.; Bhadra, R.; Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 2015, 43, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef]
- Luo, M.; He, C.; Toth, K.S.; Zhang, C.X.; Lipton, H.L. Three-dimensional structure of Theiler murine encephalomyelitis virus (BeAn strain). Proc. Natl. Acad. Sci. USA 1992, 89, 2409–2413. [Google Scholar] [CrossRef]
- Zurbriggen, A.; Hogle, J.M.; Fujinami, R.S. Alteration of Amino Acid 101 Within Capsid Protein VP-1 Changes the Pathogenicity of Theiler’s Murine Encephalomyelitis Virus. J. Exp. Med. 1989, 170, 2037–2049. [Google Scholar] [CrossRef]
- Shakeel, S.; Westerhuis, B.M.; Domanska, A.; Koning, R.I.; Matadeen, R.; Koster, A.J.; Bakker, A.Q.; Beaumont, T.; Wolthers, K.C.; Butcher, S.J. Multiple capsid-stabilizing interactions revealed in a high-resolution structure of an emerging picornavirus causing neonatal sepsis. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Hertzler, S.; Luo, M.; Lipton, H.L. Mutation of Predicted Virion Pit Residues Alters Binding of Theiler’s Murine Encephalomyelitis Virus to BHK-21 Cells. J. Virol. 2000, 74, 1994–2004. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mullapudi, E.; Nováček, J.; Pálková, L.; Kulich, P.; Lindberg, A.M.; van Kuppeveld, F.J.M.; Plevka, P. Structure and Genome Release Mechanism of the Human Cardiovirus Saffold Virus 3. J. Virol. 2016, 90, 7628–7639. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhang, R.; Liu, T.; Sun, Z.; Hu, M.; Sun, Y.; Cheng, L.; Guo, Y.; Fu, S.; Hu, J.; et al. Seneca Valley virus attachment and uncoating mediated by its receptor anthrax toxin receptor 1. Proc. Natl. Acad. Sci. USA 2018, 115, 13087–13092. [Google Scholar] [CrossRef]
- Scott, K.A.; Kotecha, A.; Seago, J.; Ren, J.; Fry, E.E.; Stuart, D.I.; Charleston, B.; Maree, F.F. SAT2 Foot-and-Mouth Disease Virus Structurally Modified for Increased Thermostability. J. Virol. 2017, 91, e02312-16. [Google Scholar] [CrossRef] [PubMed]
- Garriga, D.; Pickl-Herk, A.; Luque, D.; Wruss, J.; Castón, J.R.; Blaas, D.; Verdaguer, N. Insights into Minor Group Rhinovirus Uncoating: The X-ray Structure of the HRV2 Empty Capsid. PLoS Pathog. 2012, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zurbriggen, A.; Thomas, C.; Yamada, M.; Roos, R.P.; Fujinami, R.S. Direct evidence of a role for amino acid 101 of VP-1 in central nervous system disease in Theiler’s murine encephalomyelitis virus infection. J. Virol. 1991, 65, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Buchta, D.; Füzik, T.; Hrebík, D.; Levdansky, Y.; Sukeník, L.; Mukhamedova, L.; Moravcová, J.; Vácha, R.; Plevka, P. Enterovirus particles expel capsid pentamers to enable genome release. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Lyu, K.; Ding, J.; Han, J.; Zhang, Y.; Wu, X.; He, Y.; Qin, C.; Chen, R. Human Enterovirus 71 Uncoating Captured at Atomic Resolution. J. Virol. 2014, 88, 3114–3126. [Google Scholar] [CrossRef]
- Curry, S.; Fry, E.; Blakemore, W.; Abu-Ghazaleh, R.; Jackson, T.; King, A.; Lea, S.; Newman, J.; Stuart, D. Dissecting the Roles of VP0 Cleavage and RNA Packaging in Picornavirus Capsid Stabilization: The Structure of Empty Capsids of Foot-and-Mouth Disease Virus. J. Virol. 1997, 71, 9743–9752. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, X.; Ren, J.; Porta, C.; Wenham, H.; Ekström, J.O.; Panjwani, A.; Knowles, N.J.; Kotecha, A.; Siebert, C.A.; et al. Structure of Ljungan virus provides insight into genome packaging of this picornavirus. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Malik, N.; Kotecha, A.; Gold, S.; Asfor, A.; Ren, J.; Huiskonen, J.T.; Tuthill, T.J.; Fry, E.E.; Stuart, D.I. Structures of foot and mouth disease virus pentamers: Insight into capsid dissociation and unexpected pentamer reassociation. PLoS Pathog. 2017, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.S.; Martin, J.H. Picornavirus uncoating. J. Clin. Pathol. Mol. Pathol. 2002, 55, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.G. The canyon hypothesis. Hiding the host cell receptor attachment site on a viral surface from immune surveillance. J. Biol. Chem. 1989, 264, 14587–14590. [Google Scholar] [PubMed]
- Reddi, H.V.; Kumar, A.S.M.; Kung, A.Y.; Kallio, P.D.; Schlitt, B.P.; Lipton, H.L. Heparan Sulfate-Independent Infection Attenuates High-Neurovirulence GDVII Virus-Induced Encephalitis. J. Virol. 2004, 78, 8909–8916. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suomalainen, M.; Greber, U.F. Uncoating of non-enveloped viruses. Curr. Opin. Virol. 2013, 3, 27–33. [Google Scholar] [CrossRef]
- Bostina, M.; Levy, H.; Filman, D.J.; Hogle, J.M. Poliovirus RNA Is Released from the Capsid near a Twofold Symmetry Axis. J. Virol. 2011, 85, 776–783. [Google Scholar] [CrossRef]
- Butan, C.; Filman, D.J.; Hogle, J.M. Cryo-Electron Microscopy Reconstruction Shows Poliovirus 135S Particles Poised for Membrane Interaction and RNA Release. J. Virol. 2014, 88, 1758–1770. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, Y.; Jiang, W.; Smith, T.J.; Xu, Z.; Rossmann, M.G. Antibody-induced uncoating of human rhinovirus B14. Proc. Natl. Acad. Sci. USA 2017, 114, 8017–8022. [Google Scholar] [CrossRef]
- Liu, Y.; Sheng, J.; van Vliet, A.L.W.; Buda, G.; van Kuppeveld, F.J.M.; Rossmann, M.G. Molecular basis for the acid-initiated uncoating of human enterovirus D68. Proc. Natl. Acad. Sci. USA 2018, 115, E12209–E12217. [Google Scholar] [CrossRef]
- Shakeel, S.; Seitsonen, J.J.T.; Kajander, T.; Laurinmaki, P.; Hyypia, T.; Susi, P.; Butcher, S.J. Structural and Functional Analysis of Coxsackievirus A9 Integrin αvβ6 Binding and Uncoating. J. Virol. 2013, 87, 3943–3951. [Google Scholar] [CrossRef]
- Wang, X.; Peng, W.; Ren, J.; Hu, Z.; Xu, J.; Lou, Z.; Li, X.; Yin, W.; Shen, X.; Porta, C.; et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, J.J.T.; Shakeel, S.; Susi, P.; Pandurangan, A.P.; Sinkovits, R.S.; Hyvonen, H.; Laurinmaki, P.; Yla-Pelto, J.; Topf, M.; Hyypia, T.; et al. Structural Analysis of Coxsackievirus A7 Reveals Conformational Changes Associated with Uncoating. J. Virol. 2012, 86, 7207–7215. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, S.; Kumar, M.; Sedivy, A.; Subirats, X.; Kowalski, H.; Köhler, G.; Blaas, D. Viral Uncoating Is Directional: Exit of the Genomic RNA in a Common Cold Virus Starts with the Poly-(A) Tail at the 3′-End. PLoS Pathog. 2013, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, T.J.; Harlos, K.; Walter, T.S.; Knowles, N.J.; Groppelli, E.; David, J.; Stuart, D.I.; Fry, E.E. Equine Rhinitis A Virus and Its Low pH Empty Particle: Clues Towards an Aphthovirus Entry Mechanism? PLoS Pathog. 2009, 5, 1–11. [Google Scholar] [CrossRef][Green Version]
- Mateu, M.G. The Foot-and-mouth Disease Virion: Structure and Function. In Foot-and-Mouth Disease Virus: Current Research and Emerging Trends; Sobrino, F.S., Domingo, E., Eds.; Caister Academic Press: Norfolk, UK, 2017; pp. 61–106. ISBN 978-1-910190-52-4. [Google Scholar]
- Li, C.; Wang, J.C.Y.; Taylor, M.W.; Zlotnick, A. In Vitro Assembly of an Empty Picornavirus Capsid follows a Dodecahedral Path. J. Virol. 2012, 86, 13062–13069. [Google Scholar] [CrossRef]
- Warwicker, J. A theoretical study of the acidification of the rhinovirus capsid. FEBS Lett. 1989, 257, 403–407. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Web Server/Database | Strategy | Technique | Ref. |
|---|---|---|---|
| ROBETTA http://www.robetta.org/alascansubmit.jsp | Energy-based: | Computational alanine scanning: Interfacial residues are individually substituted to alanine. The Δ binding free energy of the complex and Δ protein stability are calculated. Hotspots are defined as residues that increase the binding free energy ≥1 kcal/mol following substitution to alanine. | [23] |
| Free energy function. | |||
| PPCheck http://caps.ncbs.res.in/ppcheck/ | Energy- and feature-based: | Energy scoring scheme: Calculates and imparts pseudoenergies to noncovalent interactions in protein–protein interfaces. These energies are weighted with features to identify hotspot residues. | [40] |
| Energy scoring, extent of spatial residue interaction, extent of energy contribution. | |||
| PredHS http://predhs.denglab.org/ | Feature- and energy-based: | Machine Learning: Euclidian and Voronoi neighbourhoods are used to weight features and generate individual residue scores. Hotspots are defined as residues with scores >0. | [41] |
| 38 sequence-, structure- and energy-based features. | |||
| KFC2 https://mitchell-web.ornl.gov/KFC_Server/upload.php | Feature-based: | Machine Learning: Various features are assessed by two models built using support vector machines. Residues which are detected as hotspots are highlighted. | [42,43] |
| Residue size, packing density, solvent accessibility, hydrophobicity, flexibility. | |||
| HotRegion/Hotpoint http://prism.ccbb.ku.edu.tr/hotregion | Feature-based: | Machine Learning: HotPoint assesses features using an empirical model and defines a residue as a hotspot if ASA values are ≤20% and contact potential values ≥18.0. HotRegion creates a network of these hotspots and highlights those which form contacts. | [44] |
| Solvent accessibility (ASA) and residue contact potential/known residue pair energies. |
| Web Server | Technique | Interactions and Cutoff Distances | Ref. |
|---|---|---|---|
| jsPISA http://www.ccp4.ac.uk/pisa | Uses seven parameters to identify interfaces. Solvation energy, binding energy, hydrophobic p-value, number and type of contacts are then determined. | hydrogen bonds; salt bridges; disulphide bonds | [46] |
| PIC http://pic.mbu.iisc.ernet.in/ | Predicts residue–residue interactions at protein–protein interfaces from the atomic coordinates of the input complex using standard widely accepted criteria. | hydrogen bonds; hydrophobic interactions (5 Å); cation-pi and ionic interactions (6 Å); disulphide bonds and aromatic–aromatic interactions bonds (4.5–7 Å); aromatic–sulphur interactions (5.3 Å) | [45] |
| Hotspot Residue | Known Function | Ref. |
|---|---|---|
| VP1: R94, W95 & V96 VP2: F176 & M178 | Reside on VP1 loop II and VP2 Puff A. Interactions between these residues are predicted to increase the stability of these loops. The substitution of A101 within the VP1 loop to tryptophan disrupted interactions between these residues and reduced virus yield and persistence. | [49,50] |
| VP1: W202, W206 & F215 VP2: Y135 & E146 | W202-Y135 form a strong hydrophobic core which stabilizes the TMEV VP1 foot-and-mouth-disease virus (FMDV) loop. E146 on VP2 puff B forms hydrophobic interactions with a non-hotspot on the FMDV loop of VP1. These interactions allow TMEV, unlike other cardioviruses, to remain stable under a broad range of pH conditions. | [49] |
| VP1: F254 VP3: R100 | Reside in receptor binding site and were predicted to be involved in binding to the TMEV co-receptor heparan sulphate. | [38] |
| VP1: V245, F246, R249, L252, F253, F254, W256, T260 & I272 | Form part of the VP1 C-terminal loop located over the receptor binding site. | [38] |
| Hotspot Residue | Corresponding Residues in Related Viruses with Known Function(s) | Ref. |
|---|---|---|
| VP1: K241 & R249 | Correspond to residues K256 and R264 in enterovirus 71 (EV-71), which have been shown to be necessary for virus replication. In vitro substitution of either residue to alanine was lethal as virus could not be recovered. | [20] |
| VP1: Y124, N204, F246 & R249 | Conserved with energetically important residues Y128, D206, W261, and R264 (VP1) that form part of the intraprotomer interfaces of EV-71 which were found in a conserved motif within the enteroviruses. | [36] |
| VP1: K241 | Corresponds to residue R202 in the VP1 protein of human parechovirus 3 (HPeV-3) that is known to be involved in interactions with the viral genome. | [51] |
| Hotspot Residue | Known Function | Ref. |
|---|---|---|
| VP1: P153 VP3: I181 | These residues are exposed at the bottom of the putative receptor binding site in TMEV. P153 was previously shown to be critical for binding the unknown glycoprotein receptor. | [52] |
| Hotspot Residue | Corresponding Residues in Related Viruses with Known Role(s) | Ref. |
|---|---|---|
| VP1: P153, VP3: N103, Q104, Q173, I181 & M222 | Conserved with residues in Saffold virus 3 (SAFV-3) that undergo conformational transitions to form a pore at the protomer–protomer interface in the expanded particle, which is thought to be involved in RNA release. | [53] |
| VP3: S16 | Corresponds to residue T47 in the VP3 protein of HPeV-3 that is known to make contacts with the RNA genome. | [51] |
| Hotspot Residue | Corresponding Residues in Related Viruses with Known Function(s) | Ref. |
|---|---|---|
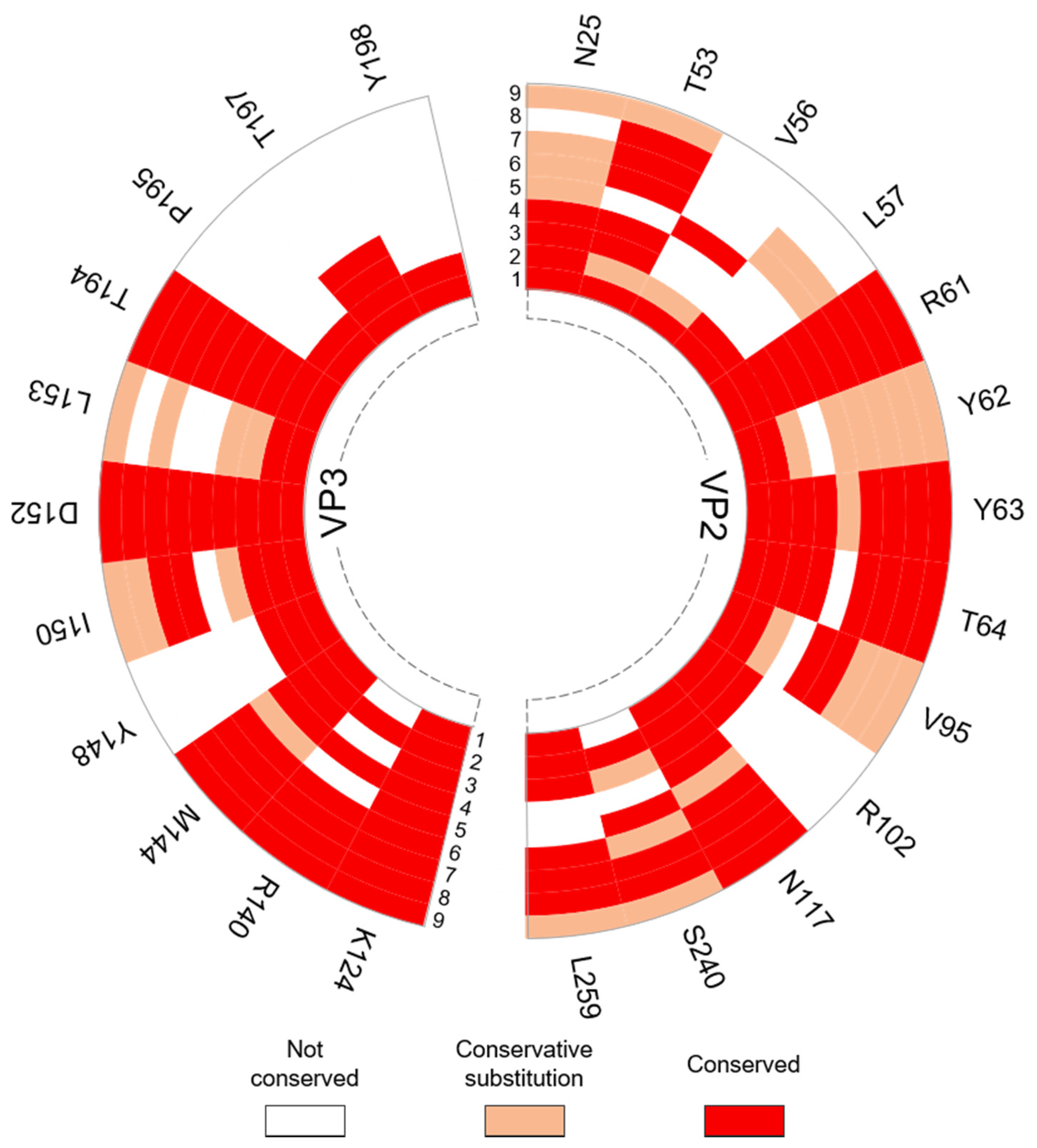
| VP2: N25, R61, Y62 (W in SVV-1), Y63, T64, V95 (A in SVV-1), R102, N117, S240 (T in SVV-1) VP3: M144, Y148, I150, D152, L153 (I in SVV-1), T194, T197 | Conserved with residues at the pentamer interfaces of the Seneca Valley virus 1 (SVV-1) mature capsid that are predicted to form interactions across the interface. | [54] |
| VP2: R61 VP3: K124, D152 & T194 | Correspond to residues R60 (VP2), R120, D148 and T190 (VP3) in FMDV, respectively, that were found to be important for virus growth. The in vitro substitution R60A was lethal as the virus could not be recovered. Residue substitutions R120A and D148A attenuated viral growth, yield and plaque size. The substitution of T194A could only be recovered after genotypic reversion. | [11] |
| VP2: Y63 | Corresponds to F62 in FMDV SAT2. Mutation of F62 to tyrosine, as seen in TMEV, increased the stability of the FMDV particle. | [55] |
| VP2: R61 & T64 VP3: M144 & D152 | Conserved with energetically important residues within conserved interacting motifs at the two-fold axes of enteroviruses. | [36] |
| VP3: M144, I150, D152 & L153 | Conserved with residues in human rhinovirus (HRV)-2 VP3 at the pentamer interface that become disordered during capsid uncoating and RNA release. | [56] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upfold, N.; Ross, C.; Tastan Bishop, Ö.; Knox, C. The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid. Viruses 2020, 12, 387. https://doi.org/10.3390/v12040387
Upfold N, Ross C, Tastan Bishop Ö, Knox C. The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid. Viruses. 2020; 12(4):387. https://doi.org/10.3390/v12040387
Chicago/Turabian StyleUpfold, Nicole, Caroline Ross, Özlem Tastan Bishop, and Caroline Knox. 2020. "The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid" Viruses 12, no. 4: 387. https://doi.org/10.3390/v12040387
APA StyleUpfold, N., Ross, C., Tastan Bishop, Ö., & Knox, C. (2020). The In Silico Prediction of Hotspot Residues that Contribute to the Structural Stability of Subunit Interfaces of a Picornavirus Capsid. Viruses, 12(4), 387. https://doi.org/10.3390/v12040387