Autophagy Induced by Simian Retrovirus Infection Controls Viral Replication and Apoptosis of Jurkat T Lymphocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Virus Stock Preparation and Viral Infection
2.2. Immunofluorescence Staining
2.3. Flow Cytometry Analysis
2.4. Western Blot Analysis and Co-Immunoprecipitation Assay
2.5. RNA Interference and Lentiviral Transduction
2.6. Realtime Quantitative RT-PCR
2.7. MTT Colorimetric Assay
2.8. Statistical Analysis
3. Results
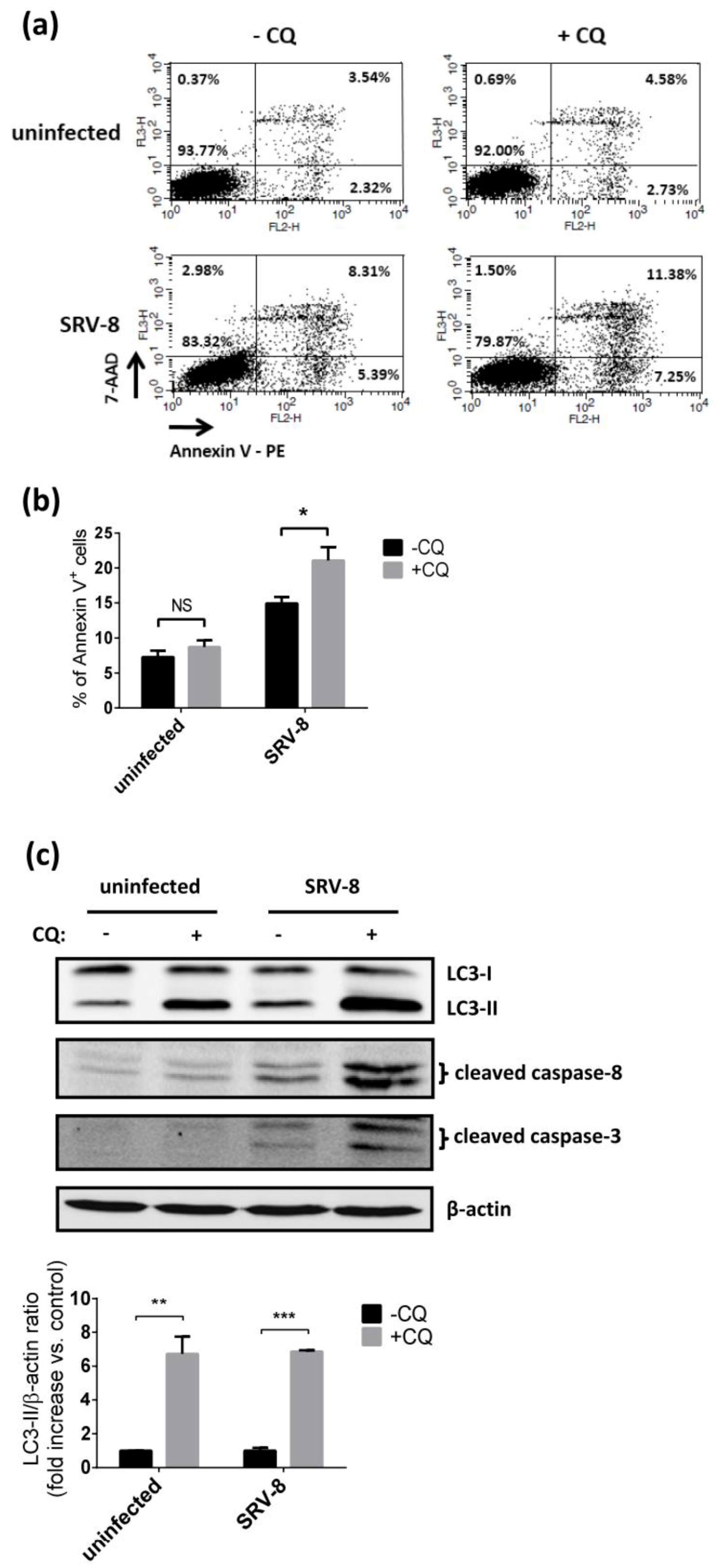
3.1. SRV-8 Infection Enhances Autophagosome Formation and Autophagic Flux in Jurkat Cells
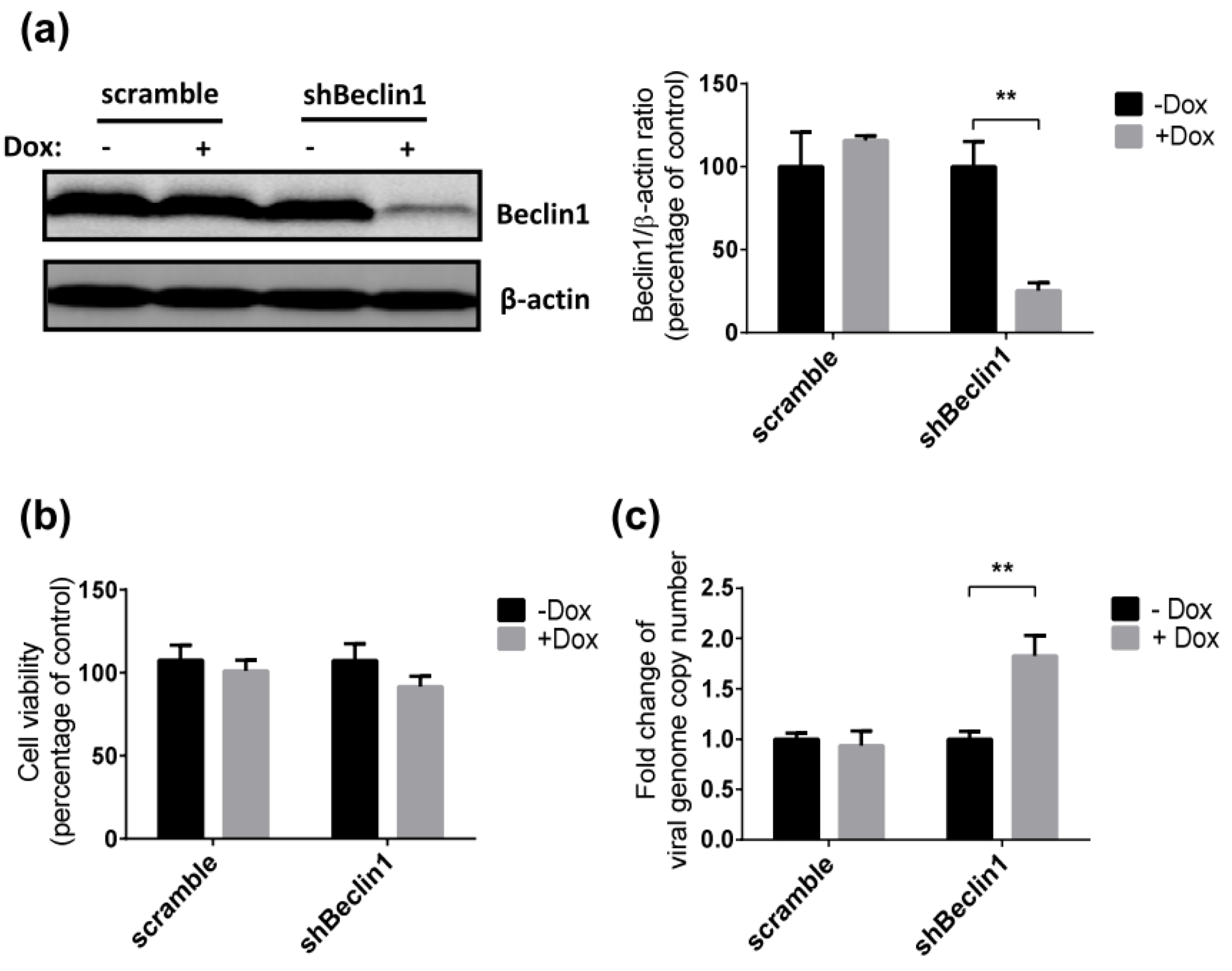
3.2. Inhibition of Autophagy Enhances SRV-8 Replication in Jurkat Cells
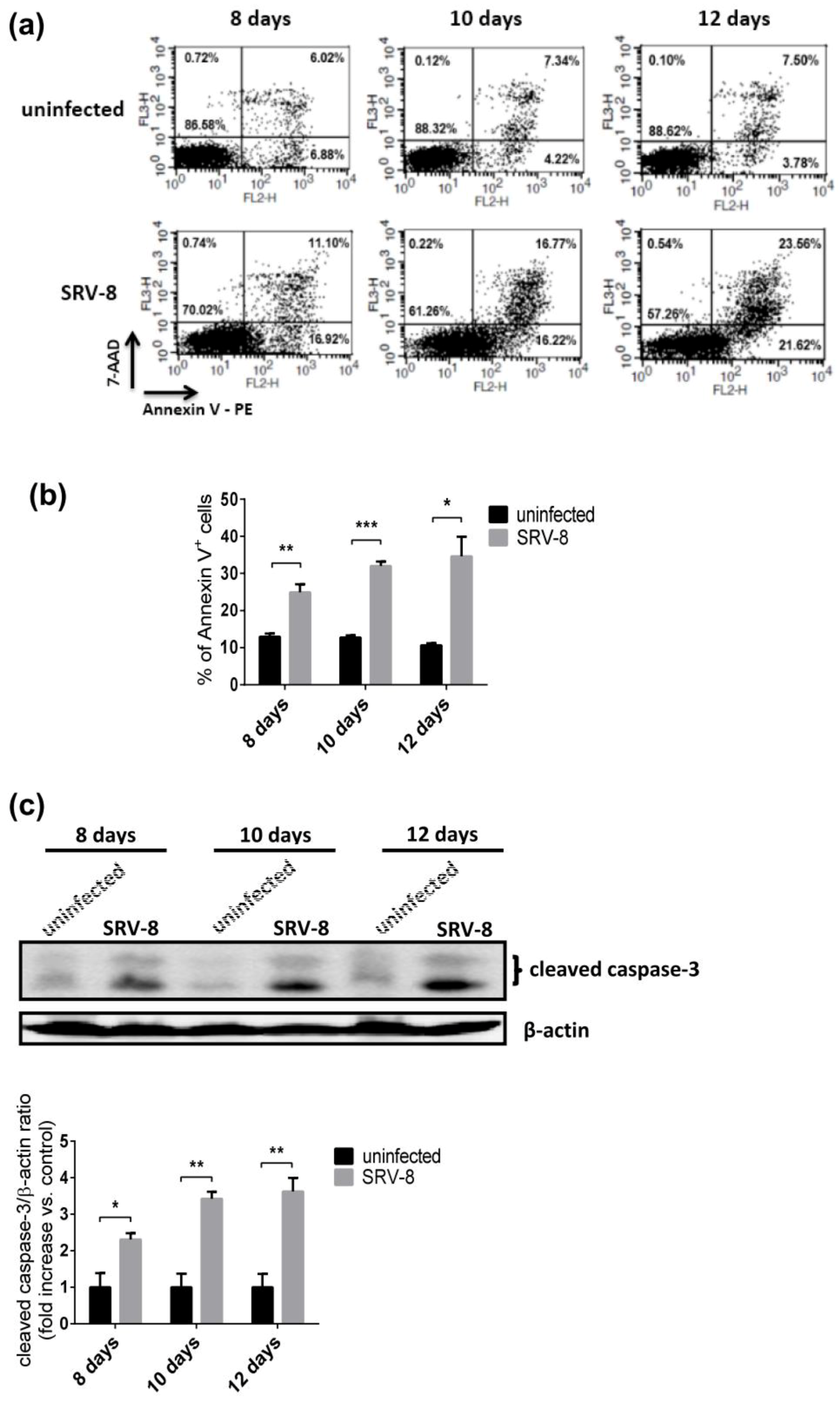
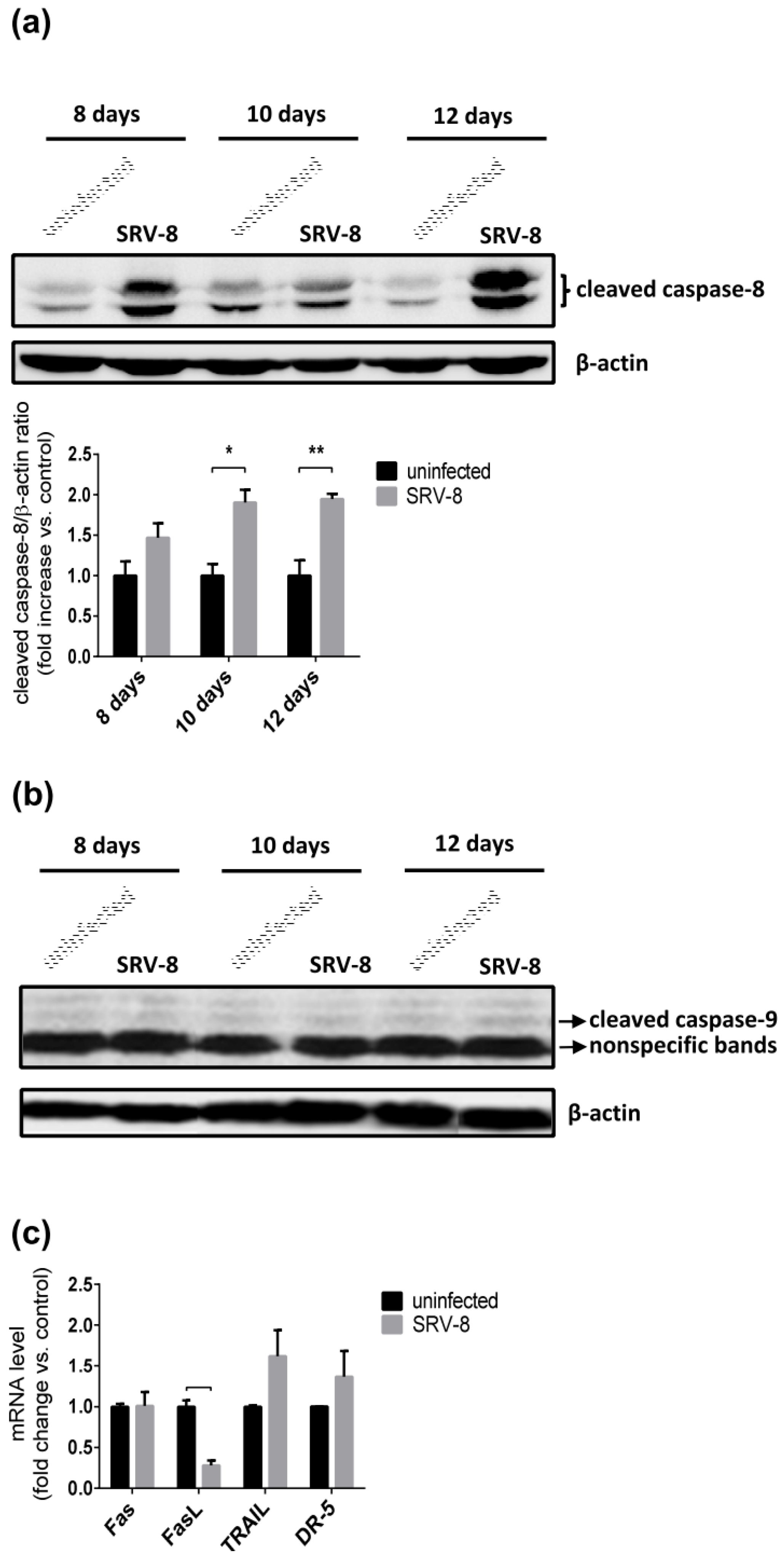
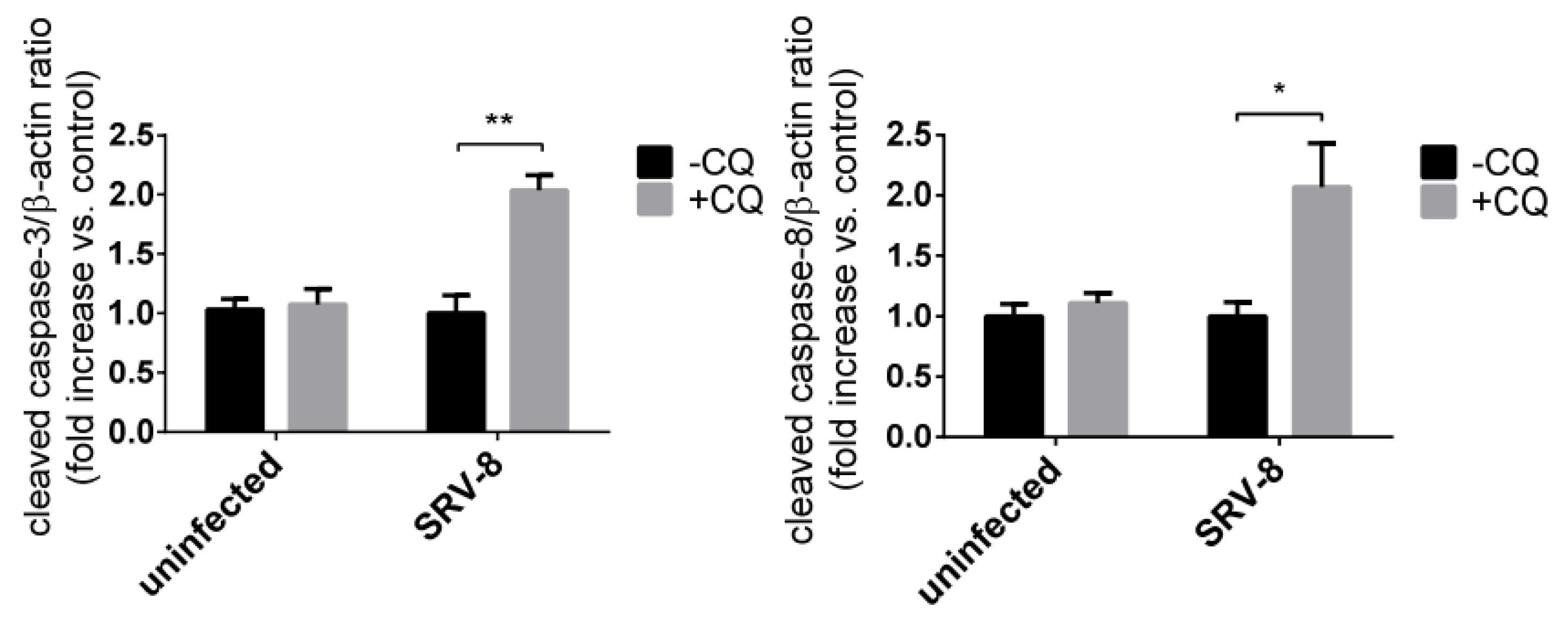
3.3. SRV-8 Infection Induces Apoptosis and Activation of Caspase-8 in Jurkat Cells
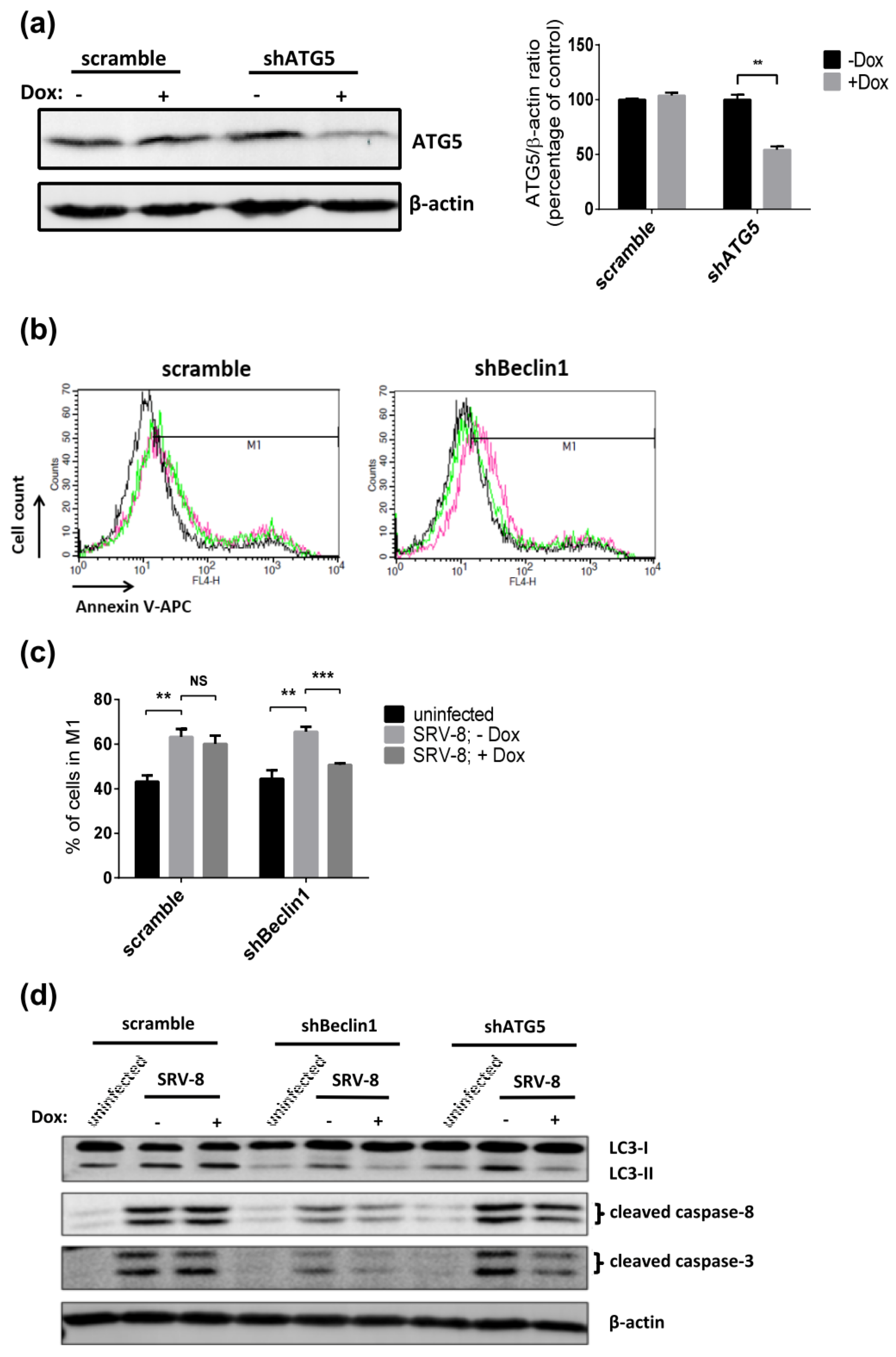
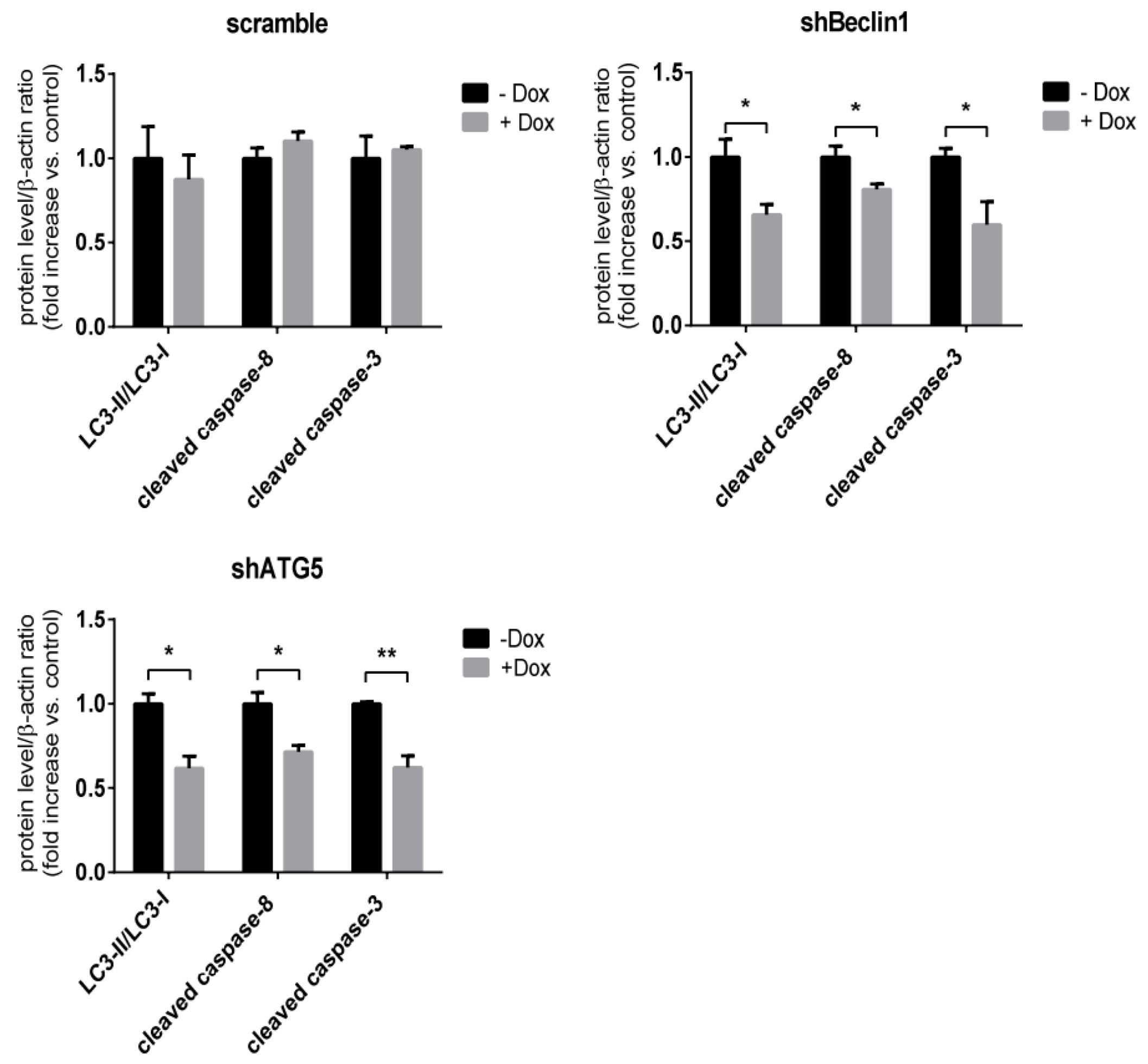
3.4. Apoptosis in SRV-8-Infected Jurkat Cells is Regulated by Autophagosome Formation
3.5. Procaspase-8 Localizes to Autophagosomes and Interacts with LC3 and p62/SQSTM1 in SRV-8-Infected Jurkat Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Dawson, P.W.; Richardson, C.D. Viral interactions with macroautophagy: A double-edged sword. Virology 2010, 402, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Ou, J.H. Autophagy in viral replication and pathogenesis. Mol. Cells 2010, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Chisari, F.V. Viruses and the autophagy machinery. Cell Cycle 2010, 9, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Chang, C.L.; Wang, P.S.; Tsai, Y.; Liu, H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar] [CrossRef]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Tan, J.; Devadas, K.; Ragupathy, V.; Takeda, K.; Zhao, J.; Hewlett, I. HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4+ T cells. Cell. Signal. 2012, 24, 1414–1419. [Google Scholar] [CrossRef]
- Alexander, D.E.; Leib, D.A. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 2008, 4, 101–103. [Google Scholar] [CrossRef]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Nain, M.; Kaur, M.; Sood, V.; Gupta, V.; Khasa, R.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014, 10, 1637–1651. [Google Scholar] [CrossRef]
- Schmid, D.; Munz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Rey-Jurado, E.; Riedel, C.A.; Gonzalez, P.A.; Bueno, S.M.; Kalergis, A.M. Contribution of autophagy to antiviral immunity. Febs Lett. 2015, 589, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Viral evasion of autophagy. Autophagy 2008, 4, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Wileman, T. Aggresomes and autophagy generate sites for virus replication. Science 2006, 312, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. How positive-strand RNA viruses benefit from autophagosome maturation. J. Virol. 2013, 87, 9966–9972. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef]
- Ren, Y.; Li, C.; Feng, L.; Pan, W.; Li, L.; Wang, Q.; Li, J.; Li, N.; Han, L.; Zheng, X.; et al. Proton Channel Activity of Influenza A Virus Matrix Protein 2 Contributes to Autophagy Arrest. J. Virol. 2016, 90, 591–598. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagy: Dual roles in life and death? Nat. Reviews. Mol. Cell Biol. 2005, 6, 505–510. [Google Scholar] [CrossRef]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis Int. J. Program. Cell Death 2008, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; He, M.X.; McLeod, I.X.; Guo, J.; Ji, D.; He, Y.W. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 2015, 11, 2335–2345. [Google Scholar] [CrossRef] [PubMed]
- Jacquin, E.; Apetoh, L. Cell-Intrinsic Roles for Autophagy in Modulating CD4 T Cell Functions. Front. Immunol. 2018, 9, 1023. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef]
- Gou, H.; Zhao, M.; Fan, S.; Yuan, J.; Liao, J.; He, W.; Xu, H.; Chen, J. Autophagy induces apoptosis and death of T lymphocytes in the spleen of pigs infected with CSFV. Sci. Rep. 2017, 7, 13577. [Google Scholar] [CrossRef]
- Li, M.; Li, J.; Zeng, R.; Yang, J.; Liu, J.; Zhang, Z.; Song, X.; Yao, Z.; Ma, C.; Li, W.; et al. Respiratory Syncytial Virus Replication Is Promoted by Autophagy-Mediated Inhibition of Apoptosis. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis Int. J. Program. Cell Death 2014, 19, 555–566. [Google Scholar] [CrossRef]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal 2014, 26, 549–555. [Google Scholar] [CrossRef]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef]
- Laussmann, M.A.; Passante, E.; Dussmann, H.; Rauen, J.A.; Wurstle, M.L.; Delgado, M.E.; Devocelle, M.; Prehn, J.H.; Rehm, M. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011, 18, 1584–1597. [Google Scholar] [CrossRef]
- Pan, J.A.; Ullman, E.; Dou, Z.; Zong, W.X. Inhibition of protein degradation induces apoptosis through a microtubule-associated protein 1 light chain 3-mediated activation of caspase-8 at intracellular membranes. Mol. Cell. Biol. 2011, 31, 3158–3170. [Google Scholar] [CrossRef] [PubMed]
- Buchen-Osmond, C. The universal virus database ICTVdB. Comput. Sci. Eng. 2003, 5, 16–25. [Google Scholar] [CrossRef]
- Montiel, N.A. An updated review of simian betaretrovirus (SRV) in macaque hosts. J. Med. Primatol. 2010, 39, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, G.; Lerche, N.W.; Lowenstine, L.J.; Lackner, A.A.; Osborn, K.G.; Gardner, M.B.; Marx, P.A. Induction of simian acquired immune deficiency syndrome (SAIDS) with a molecular clone of a type D SAIDS retrovirus. J. Virol. 1987, 61, 3066–3071. [Google Scholar] [CrossRef] [PubMed]
- Maul, D.H.; Lerche, N.W.; Osborn, K.G.; Marx, P.A.; Zaiss, C.; Spinner, A.; Kluge, J.D.; MacKenzie, M.R.; Lowenstine, L.J.; Bryant, M.L.; et al. Pathogenesis of simian AIDS in rhesus macaques inoculated with the SRV-1 strain of type D retrovirus. Am. J. Vet. Res. 1986, 47, 863–868. [Google Scholar]
- Osborn, K.G.; Prahalada, S.; Lowenstine, L.J.; Gardner, M.B.; Maul, D.H.; Henrickson, R.V. The pathology of an epizootic of acquired immunodeficiency in rhesus macaques. Am. J. Pathol. 1984, 114, 94–103. [Google Scholar]
- MacKenzie, M.; Lowenstine, L.; Lalchandani, R.; Lerche, N.; Osborn, K.; Spinner, A.; Bleviss, M.; Hendrickson, R.; Gardner, M. Hematologic abnormalities in simian acquired immune deficiency syndrome. Lab. Anim. Sci. 1986, 36, 14–19. [Google Scholar]
- Maul, D.H.; Zaiss, C.P.; MacKenzie, M.R.; Shiigi, S.M.; Marx, P.A.; Gardner, M.B. Simian retrovirus D serogroup 1 has a broad cellular tropism for lymphoid and nonlymphoid cells. J. Virol. 1988, 62, 1768–1773. [Google Scholar] [CrossRef]
- Lackner, A.A.; Rodriguez, M.H.; Bush, C.E.; Munn, R.J.; Kwang, H.S.; Moore, P.F.; Osborn, K.G.; Marx, P.A.; Gardner, M.B.; Lowenstine, L.J. Distribution of a macaque immunosuppressive type D retrovirus in neural, lymphoid, and salivary tissues. J. Virol. 1988, 62, 2134–2142. [Google Scholar] [CrossRef]
- Guzman, R.E.; Kerlin, R.L.; Zimmerman, T.E. Histologic lesions in cynomolgus monkeys (Macaca fascicularis) naturally infected with simian retrovirus type D: Comparison of seropositive, virus-positive, and uninfected animals. Toxicol. Pathol. 1999, 27, 672–677. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Murrell, C.K.; Auda, G.; Almond, N.; Rose, N.J. Early immunopathology events in simian retrovirus, type 2 infections prior to the onset of disease. Virology 2011, 413, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Zao, C.L.; Tomanek, L.; Cooke, A.; Berger, R.; Yang, L.; Xie, C.; Chen, S.; Shi, C.; Rong, R. A novel simian retrovirus subtype discovered in cynomolgus monkeys (Macaca fascicularis). J. Gen. Virol. 2016, 97, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, R.; Okamoto, M.; Sakaguchi, S.; Nakagawa, S.; Miura, T.; Hirai, H.; Miyazawa, T. Simian retrovirus 4 induces lethal acute thrombocytopenia in Japanese macaques. J. Virol. 2015, 89, 3965–3975. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marracci, G.H.; Avery, N.A.; Shiigi, S.M.; Couch, G.; Palmer, H.; Pilcher, K.Y.; Nichols, H.; Hallick, L.M.; Axthelm, M.K.; Machida, C.A. Molecular cloning and cell-specific growth characterization of polymorphic variants of type D serogroup 2 simian retroviruses. Virology 1999, 261, 43–58. [Google Scholar] [CrossRef]
- Togami, H.; Shimura, K.; Okamoto, M.; Yoshikawa, R.; Miyazawa, T.; Matsuoka, M. Comprehensive in vitro analysis of simian retrovirus type 4 susceptibility to antiretroviral agents. J. Virol. 2013, 87, 4322–4329. [Google Scholar] [CrossRef]
- Wei, Z.; Panneerdoss, S.; Timilsina, S.; Zhu, J.; Mohammad, T.A.; Lu, Z.L.; de Magalhaes, J.P.; Chen, Y.; Rong, R. Topological Characterization of Human and Mouse m(5)C Epitranscriptome Revealed by Bisulfite Sequencing. Int. J. Genom. 2018, 2018, 1351964. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Daniel, M.D.; King, N.W.; Letvin, N.L.; Hunt, R.D.; Sehgal, P.K.; Desrosiers, R.C. A new type D retrovirus isolated from macaques with an immunodeficiency syndrome. Science 1984, 223, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef]
- Yetz, J.M.; Letvin, N.L. Macaque monkey type D retrovirus replicates in vitro in a distinct subpopulation of B lymphocytes. J. Gen. Virol. 1987, 68 (Pt 2), 573–576. [Google Scholar] [CrossRef]
- Wang, X.; Tan, J.; Biswas, S.; Zhao, J.; Devadas, K.; Ye, Z.; Hewlett, I. Pandemic Influenza A (H1N1) Virus Infection Increases Apoptosis and HIV-1 Replication in HIV-1 Infected Jurkat Cells. Viruses 2016, 8, 33. [Google Scholar] [CrossRef]
- Okamoto, M.; Miyazawa, T.; Morikawa, S.; Ono, F.; Nakamura, S.; Sato, E.; Yoshida, T.; Yoshikawa, R.; Sakai, K.; Mizutani, T.; et al. Emergence of infectious malignant thrombocytopenia in Japanese macaques (Macaca fuscata) by SRV-4 after transmission to a novel host. Sci. Rep. 2015, 5, 8850. [Google Scholar] [CrossRef]
- Jordan, T.X.; Randall, G. Manipulation or capitulation: Virus interactions with autophagy. Microbes Infect. 2012, 14, 126–139. [Google Scholar] [CrossRef]
- Talloczy, Z.; Virgin, H.W.t.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef]
- Wileman, T. Autophagy as a defence against intracellular pathogens. Essays Biochem. 2013, 55, 153–163. [Google Scholar] [CrossRef]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef]
- Baehrecke, E.H. Autophagic programmed cell death in Drosophila. Cell Death Differ. 2003, 10, 940–945. [Google Scholar] [CrossRef]
- Debnath, J.; Baehrecke, E.H.; Kroemer, G. Does autophagy contribute to cell death? Autophagy 2005, 1, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Okamoto, K.; Yu, C.; Sinicrope, F.A. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J. Biol. Chem. 2013, 288, 33654–33666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Gong, J.L.; Xing, T.Y.; Zheng, S.P.; Ding, W. Autophagy protein p62/SQSTM1 is involved in HAMLET-induced cell death by modulating apotosis in U87MG cells. Cell Death Dis. 2013, 4, e550. [Google Scholar] [CrossRef]
- Tang, Z.; Takahashi, Y.; Chen, C.; Liu, Y.; He, H.; Tsotakos, N.; Serfass, J.M.; Gebru, M.T.; Chen, H.; Young, M.M.; et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 2017, 24, 2127–2138. [Google Scholar] [CrossRef]
- Pyo, J.O.; Jang, M.H.; Kwon, Y.K.; Lee, H.J.; Jun, J.I.; Woo, H.N.; Cho, D.H.; Choi, B.; Lee, H.; Kim, J.H.; et al. Essential roles of Atg5 and FADD in autophagic cell death: Dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005, 280, 20722–20729. [Google Scholar] [CrossRef]
- Jung, K.T.; Oh, S.H. Polyubiquitination of p62/SQSTM1 is a prerequisite for Fas/CD95 aggregation to promote caspase-dependent apoptosis in cadmium-exposed mouse monocyte RAW264.7 cells. Sci. Rep. 2019, 9, 12240. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.; Yang, L.; Zhang, Q.; Meng, J.; Lu, Z.-L.; Rong, R. Autophagy Induced by Simian Retrovirus Infection Controls Viral Replication and Apoptosis of Jurkat T Lymphocytes. Viruses 2020, 12, 381. https://doi.org/10.3390/v12040381
Zhu J, Yang L, Zhang Q, Meng J, Lu Z-L, Rong R. Autophagy Induced by Simian Retrovirus Infection Controls Viral Replication and Apoptosis of Jurkat T Lymphocytes. Viruses. 2020; 12(4):381. https://doi.org/10.3390/v12040381
Chicago/Turabian StyleZhu, Jingting, Lingyan Yang, Qibo Zhang, Jia Meng, Zhi-Liang Lu, and Rong Rong. 2020. "Autophagy Induced by Simian Retrovirus Infection Controls Viral Replication and Apoptosis of Jurkat T Lymphocytes" Viruses 12, no. 4: 381. https://doi.org/10.3390/v12040381
APA StyleZhu, J., Yang, L., Zhang, Q., Meng, J., Lu, Z.-L., & Rong, R. (2020). Autophagy Induced by Simian Retrovirus Infection Controls Viral Replication and Apoptosis of Jurkat T Lymphocytes. Viruses, 12(4), 381. https://doi.org/10.3390/v12040381