Proteome Analysis in a Mammalian Cell Line Reveals that PLK2 Is Involved in Avian Metapneumovirus Type C (aMPV/C)-Induced Apoptosis

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus Infection and Determination
2.2. Protein Extraction, Digestion and Labeling with iTRAQ Reagents
2.3. SCX Chromatography and LC-MS Analysis
2.4. Data Analysis
2.5. Knockdown of PLK2 by RNA Interference (RNAi)
2.6. Western Blot Analysis
2.7. Apoptosis Analysis
2.8. Reactive Oxygen Species Measurement
2.9. Measurement of Virus Release in Vero Cells
3. Results
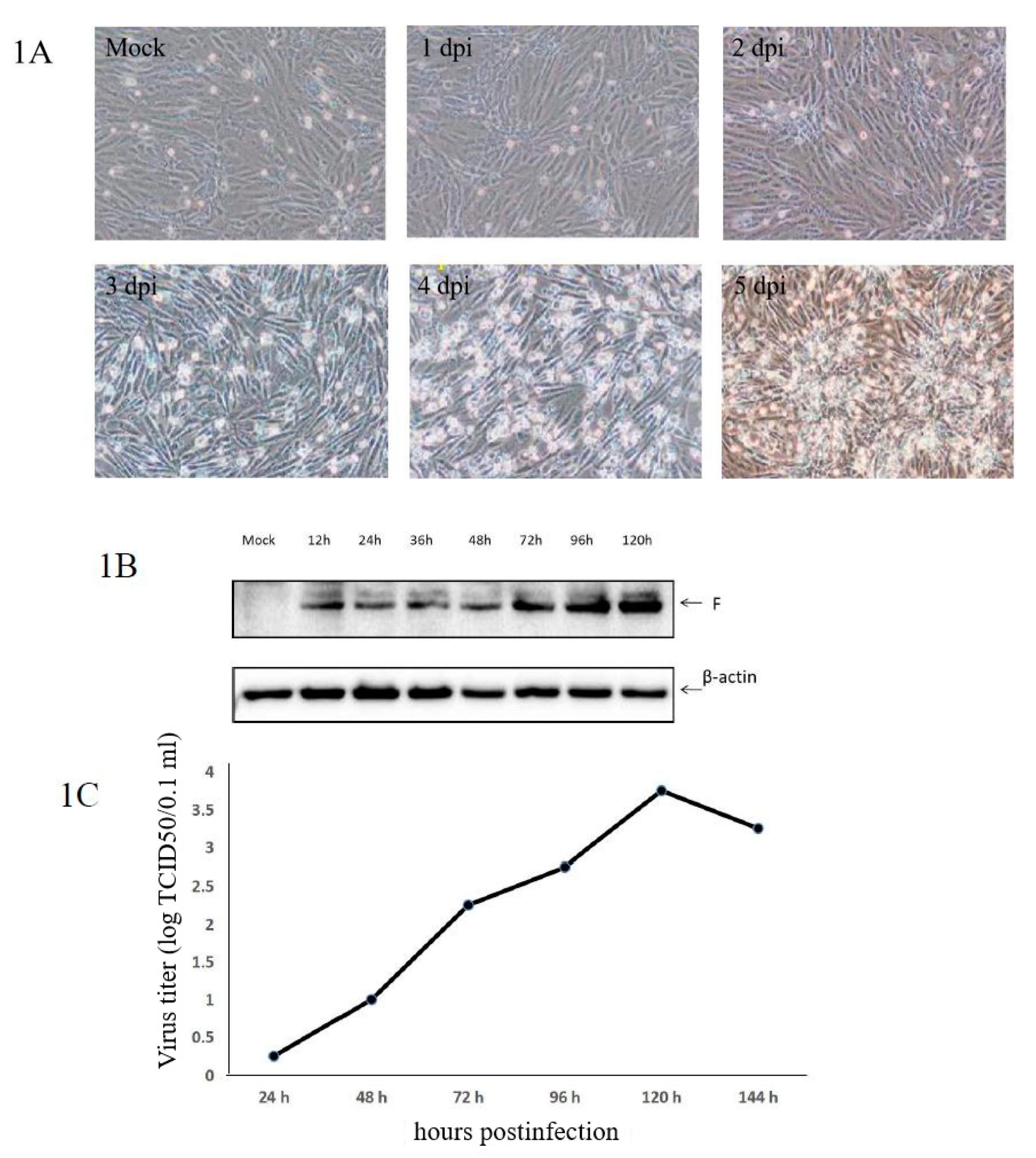
3.1. Confirmation of aMPV/C JC Propagation in Vero Cells
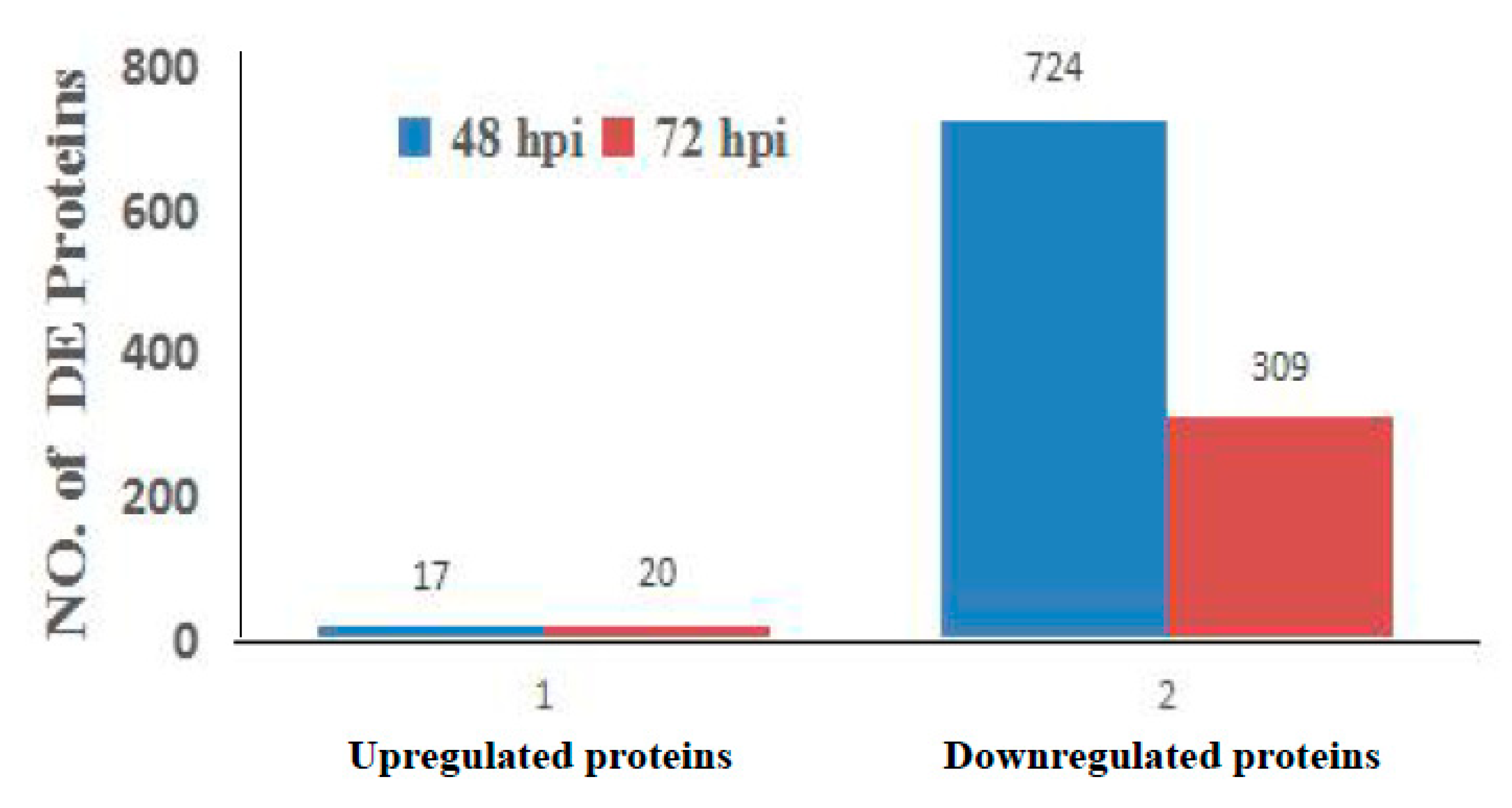
3.2. Analysis of Differentially Regulated Proteins by iTRAQ-Coupled LC-MS
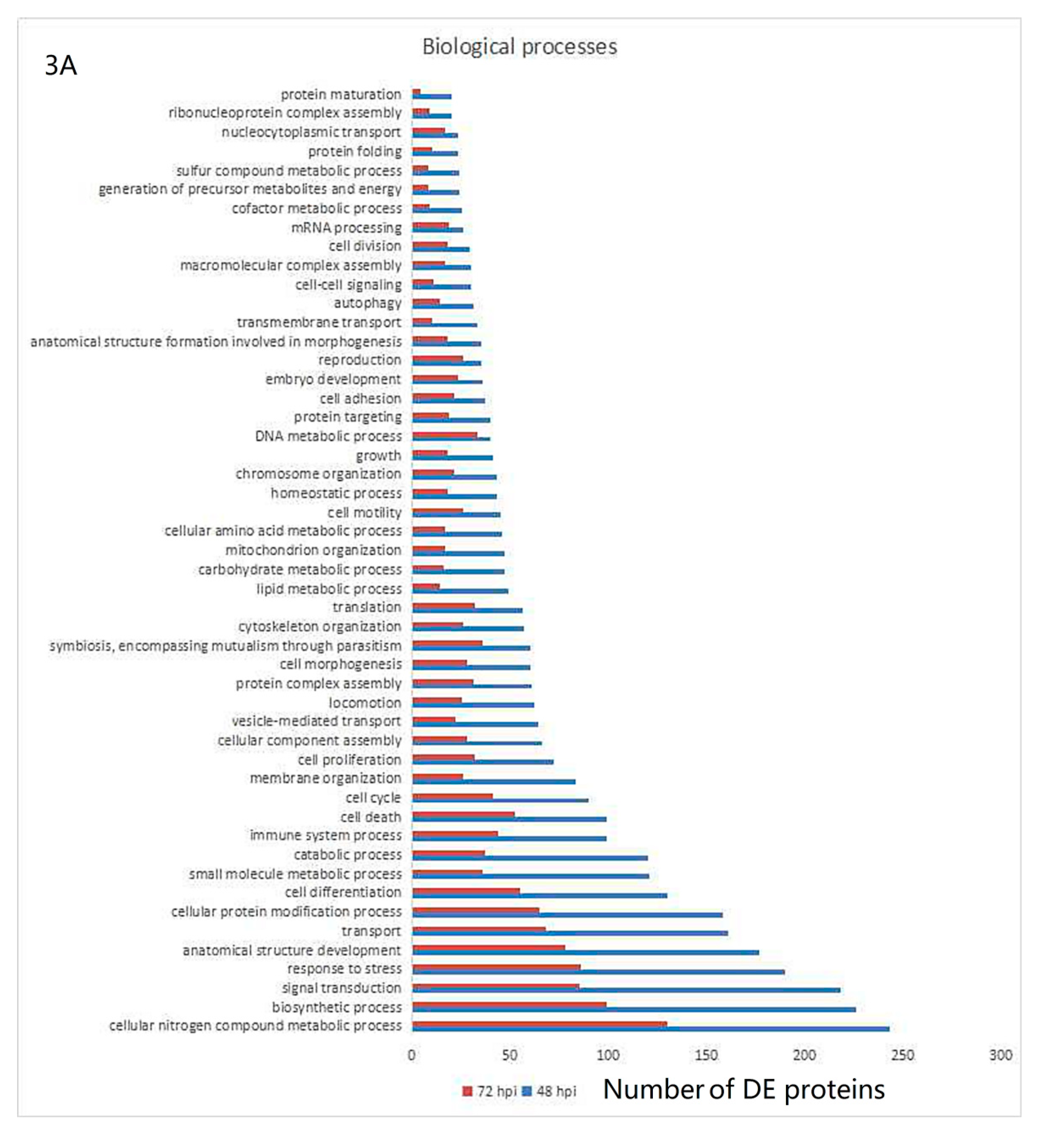
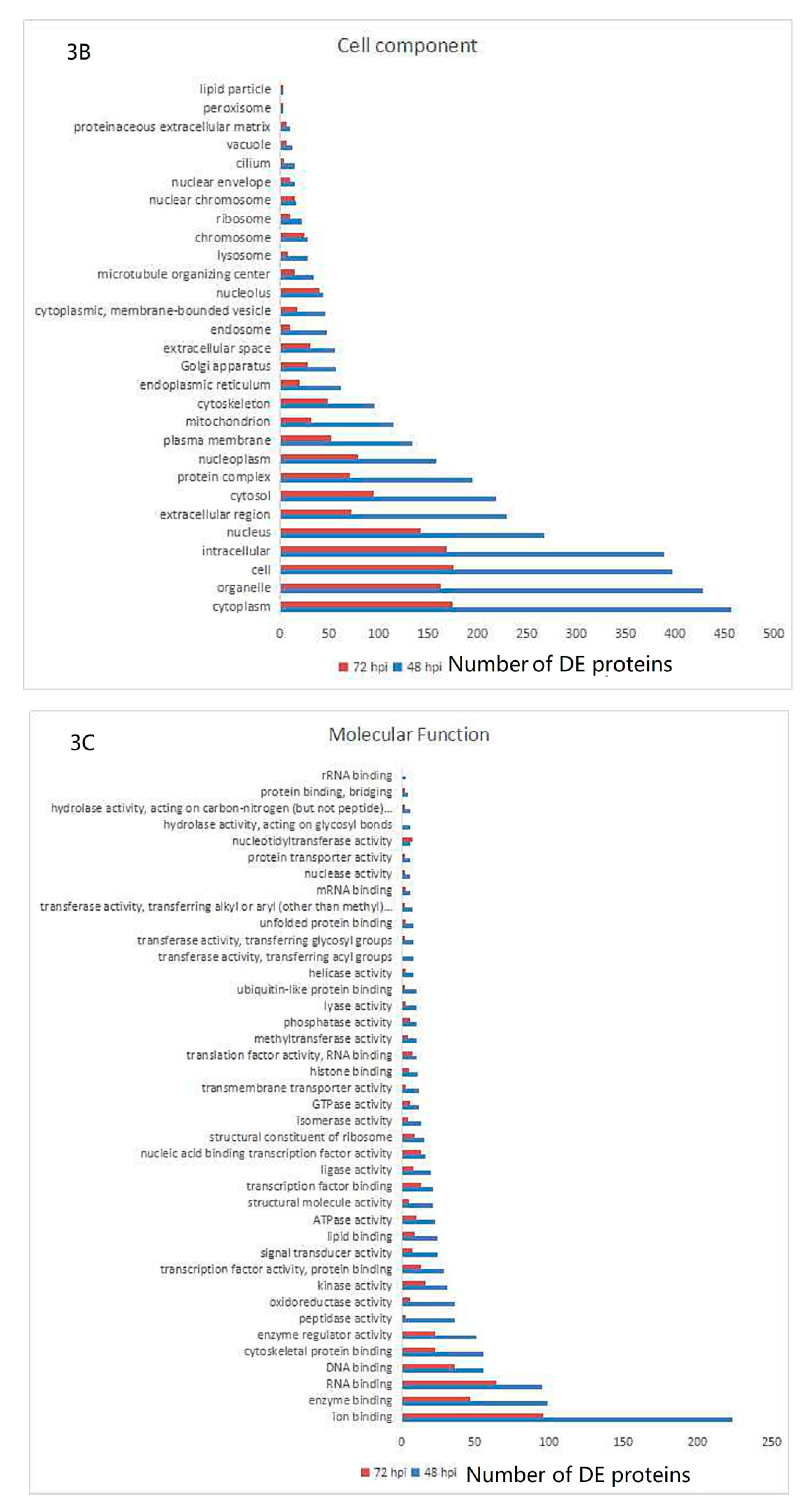
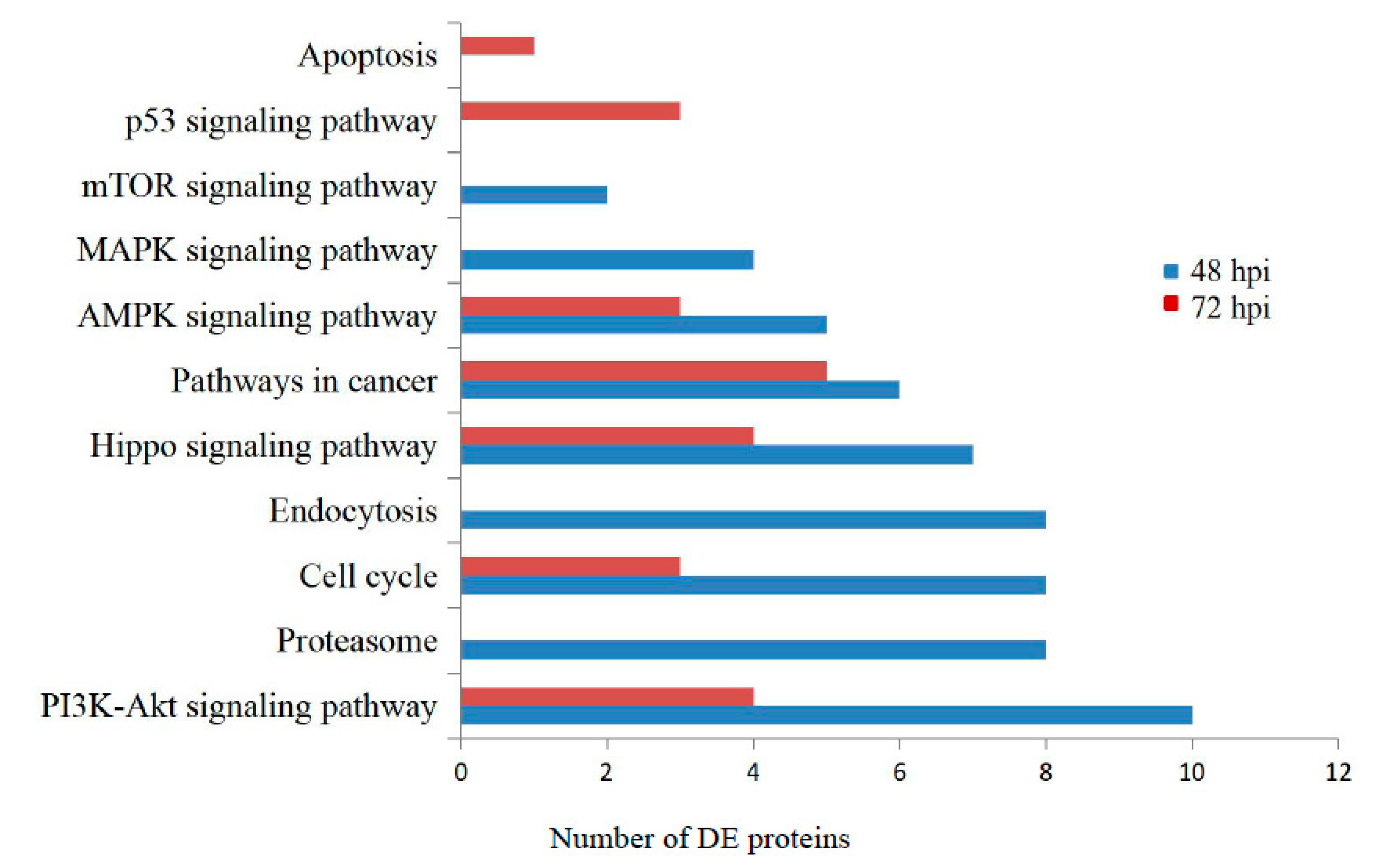
3.3. Gene Ontology and KEGG Pathway Analysis
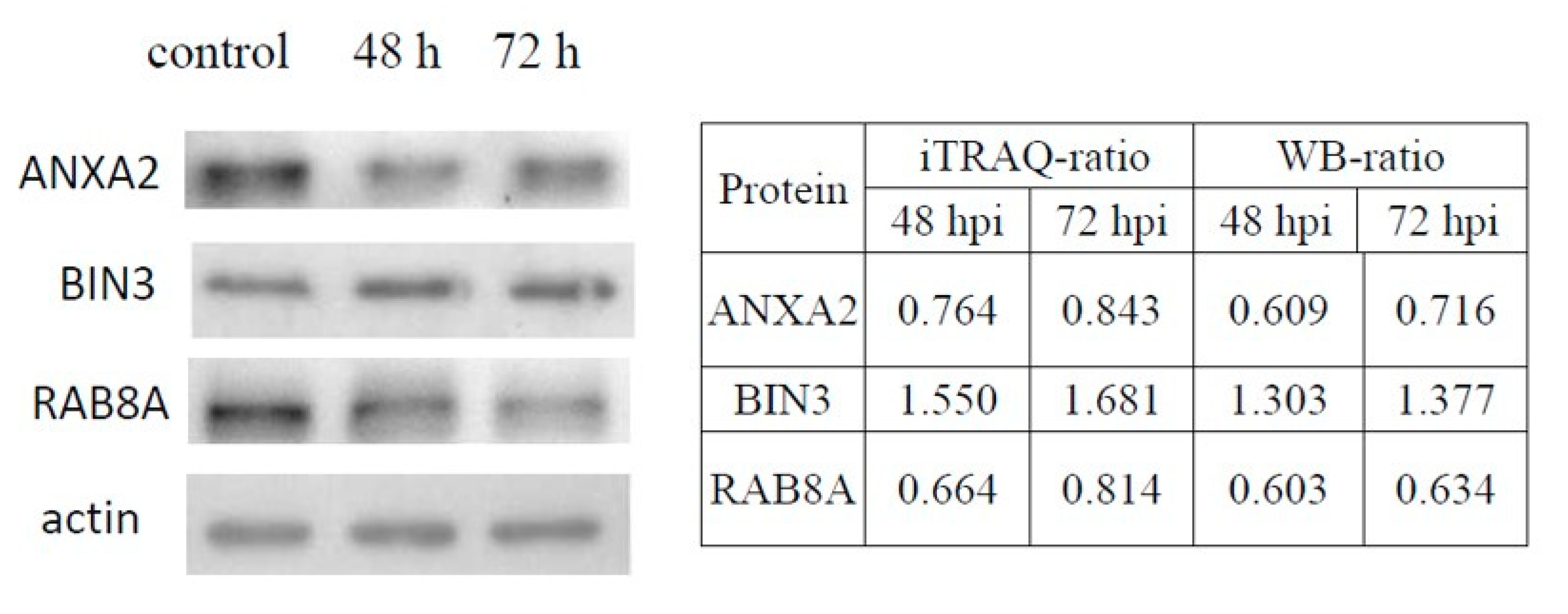
3.4. Validation of DE Proteins by Western Blotting
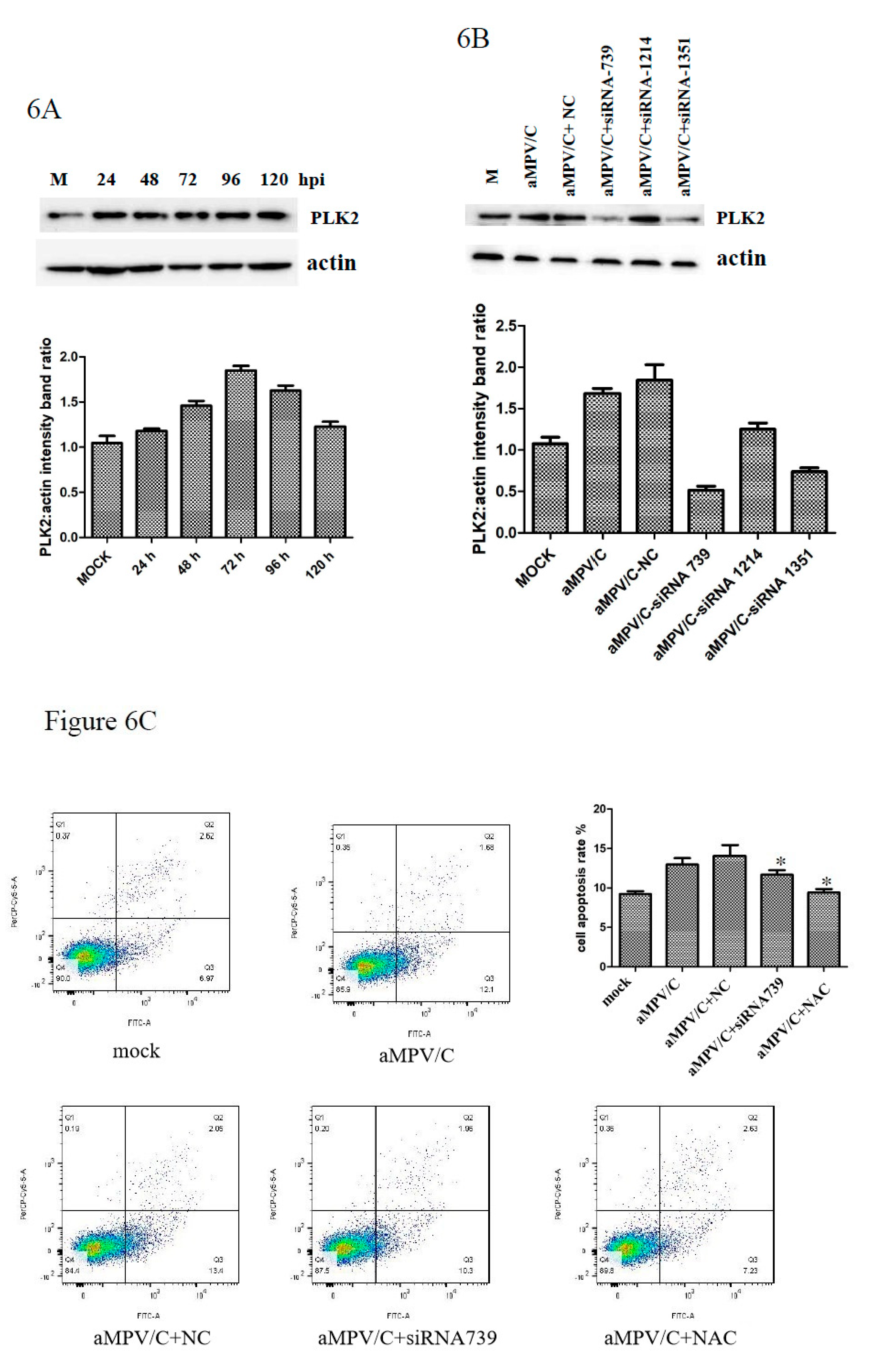
3.5. PLK2 Expression Is Upregulated and Involved in Apoptosis Induction in aMPV/C-Infected Cells
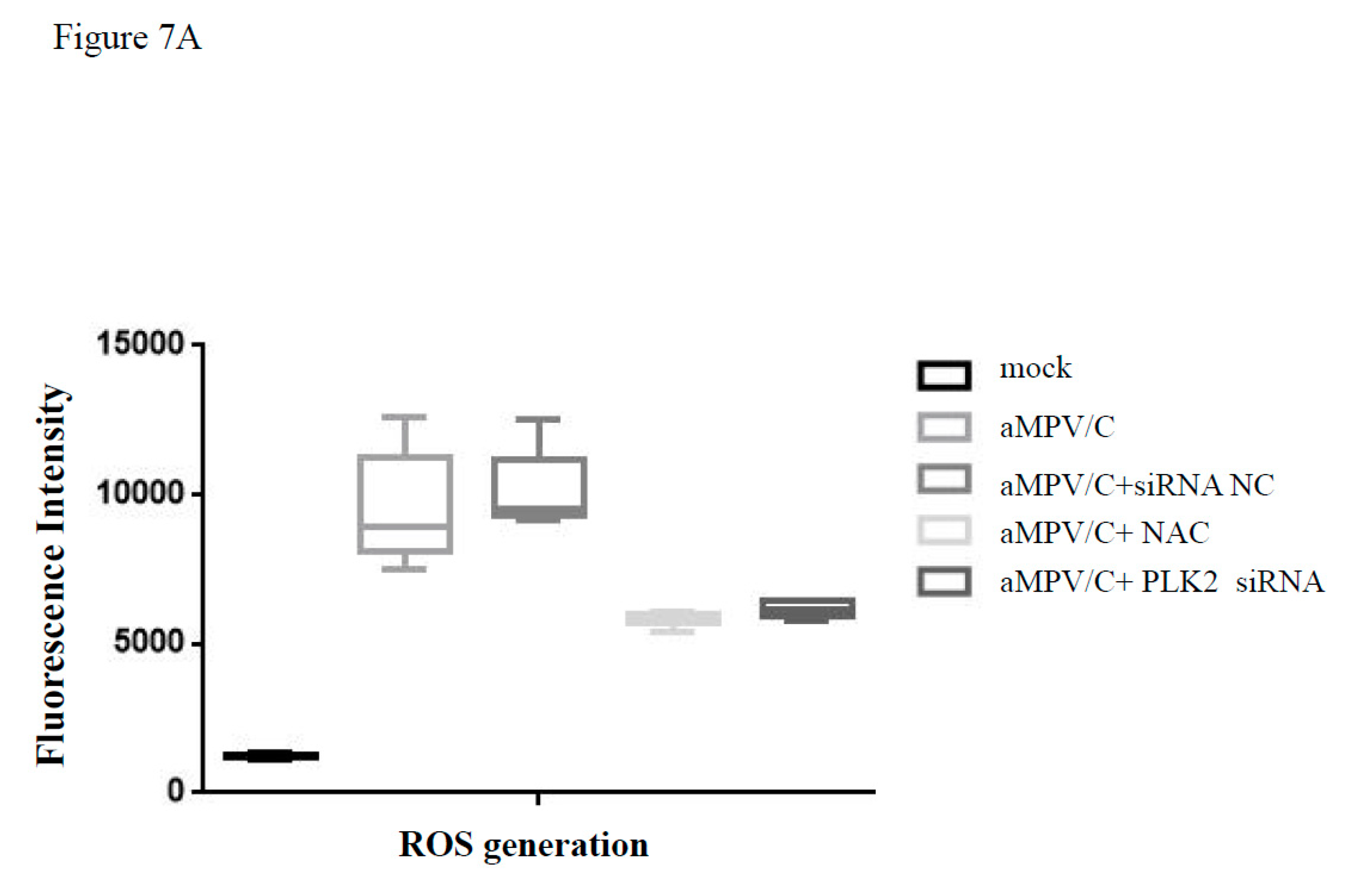
3.6. Silencing PLK2 Decreases aMPV/C-Induced ROS, Induction of Apoptosis, and Virus Release
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- McDougall, J.S.; Cook, J.K. Turkey rhinotracheitis: Preliminary investigations. Vet. Rec. 1986, 118, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K. Avian pneumovirus infections of turkeys and chickens. Vet. J. 2000, 160, 118–125. [Google Scholar] [CrossRef]
- Dilovski, M.; Ognianov, D. Isolation and identification of a virus causing abortions and stillbirths in swine (Preliminary report)]. Vet.-Meditsinski Nauk. 1975, 12, 52–53. [Google Scholar]
- Bayon-Auboyer, M.H.; Arnauld, C.; Toquin, D.; Eterradossi, N. Nucleotide sequences of the F, L and G protein genes of two non-A/non-B avian pneumoviruses (APV) reveal a novel APV subgroup. J. Gen. Virol. 2000, 81 Pt 11, 2723–2733. [Google Scholar] [CrossRef]
- Govindarajan, D.; Samal, S.K. Sequence analysis of the large polymerase (L) protein of the US strain of avian metapneumovirus indicates a close resemblance to that of the human metapneumovirus. Virus Res. 2004, 105, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Yunus, A.S.; Govindarajan, D.; Huang, Z.; Samal, S.K. Deduced amino acid sequence of the small hydrophobic protein of US avian pneumovirus has greater identity with that of human metapneumovirus than those of non-US avian pneumoviruses. Virus Res. 2003, 93, 91–97. [Google Scholar] [CrossRef]
- Brown, P.A.; Lemaitre, E.; Briand, F.X.; Courtillon, C.; Guionie, O.; Allée, C.; Toquin, D.; Bayon-Auboyer, M.H.; Jestin, V.; Eterradossi, N. Molecular comparisons of full length metapneumovirus (MPV) genomes, including newly determined French AMPV-C and -D isolates, further supports possible subclassification within the MPV genus. PLoS ONE 2014, 9, e102740. [Google Scholar] [CrossRef]
- Toquin, D.; Guionie, O.; Jestin, V.; Zwingelstein, F.; Allee, C.; Eterradossi, N. European and American subgroup C isolates of avian metapneumovirus belong to different genetic lineages. Virus Res. 2006, 32, 97–103. [Google Scholar] [CrossRef]
- Sun, S.; Chen, F.; Cao, S.; Liu, J.; Lei, W.; Li, G.; Song, Y.; Lu, J.; Liu, C.; Qin, J.; et al. Isolation and characterization of a subtype C avian metapneumovirus circulating in Muscovy ducks in China. Vet. Res. 2014, 45, 74. [Google Scholar] [CrossRef]
- Turpin, E.A.; Stallknecht, D.E.; Slemons, R.D.; Zsak, L.; Swayne, D.E. Evidence of avian metapneumovirus subtype C infection of wild birds in Georgia, South Carolina, Arkansas and Ohio, USA. Avian Pathol. 2008, 37, 343–351. [Google Scholar] [CrossRef]
- Wei, L.; Zhu, S.; Yan, X.; Wang, J.; Zhang, C.; Liu, S.; She, R.; Hu, F.; Quan, R.; Liu, J. Avian metapneumovirus subgroup C infection in chickens, China. Emerg. Infect. Dis. 2013, 19, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhu, S.; She, R.; Hu, F.; Wang, J.; Yan, X.; Zhang, C.; Liu, S.; Quan, R.; Li, Z.; et al. Viral replication and lung lesions in BALB/c mice experimentally inoculated with avian metapneumovirus subgroup C isolated from chickens. PLoS ONE 2014, 9, e92136. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Wei, L.; Zhu, S.; Wang, J.; Quan, R.; Li, Z.; Liu, J. Avian metapneumovirus subgroup C induces autophagy through the ATF6 UPR pathway. Autophagy 2017, 13, 1709–1721. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Gao, Z.; Wang, X.; Gu, M.; Liang, Y.; Liu, X.; Hu, S.; Liu, H.; Liu, W.; Chen, S.; et al. iTRAQ-based quantitative proteomics reveals important host factors involved in the high pathogenicity of the H5N1 avian influenza virus in mice. Med. Microbiol. Immunol. 2017, 206, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Li, X.; Niu, D.; Chen, W.N. Protein profile in HBx transfected cells: A comparative iTRAQ-coupled 2D LC-MS/MS analysis. J. Proteom. 2010, 73, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Zhang, Y.; Liu, H.; Ning, P. Proteome profile of swine testicular cells infected with porcine transmissible gastroenteritis coronavirus. PLoS ONE 2014, 9, e110647. [Google Scholar] [CrossRef]
- Liu, J.; Bai, J.; Zhang, L.; Hou, C.; Li, Y.; Jiang, P. Proteomic alteration of PK-15 cells after infection by porcine circovirus type 2. Virus Genes 2014, 49, 400–416. [Google Scholar] [CrossRef]
- Zhou, N.; Fan, C.; Liu, S.; Zhou, J.; Jin, Y.; Zheng, X.; Wang, Q.; Liu, J.; Yang, H.; Gu, J.; et al. Cellular proteomic analysis of porcine circovirus type 2 and classical swine fever virus coinfection in porcine kidney-15 cells using isobaric tags for relative and absolute quantitation-coupled LC-MS/MS. Electrophoresis 2017, 38, 1276–1291. [Google Scholar] [CrossRef]
- Lu, Q.; Bai, J.; Zhang, L.; Liu, J.; Jiang, Z.; Michal, J.J.; He, Q.; Jiang, P. Two-dimensional liquid chromatography-tandem mass spectrometry coupled with isobaric tags for relative and absolute quantification (iTRAQ) labeling approach revealed first proteome profiles of pulmonary alveolar macrophages infected with porcine reproductive and respiratory syndrome virus. J. Proteome Res. 2012, 11, 2890–2903. [Google Scholar]
- Zeng, S.; Zhang, H.; Ding, Z.; Luo, R.; An, K.; Liu, L.; Bi, J.; Chen, H.; Xiao, S.; Fang, L. Proteome analysis of porcine epidemic diarrhea virus (PEDV)-infected Vero cells. Proteomics 2015, 15, 1819–1828. [Google Scholar] [CrossRef]
- Lin, H.; Li, B.; Chen, L.; Ma, Z.; He, K.; Fan, H. Differential protein analysis of IPEC-J2 cells infected with porcine epidemic diarrhea virus pandemic and classical strains elucidates the pathogenesis of infection. J. Proteome Res. 2017, 16, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jang, C.; Lee, K.A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Donohue, P.J.; Alberts, G.F.; Guo, Y.; Winkles, J.A. Identification by targeted differential display of an immediate early gene encoding a putative serine/threonine kinase. J. Biol. Chem. 1995, 270, 10351–10357. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Charron, J.; Erikson, R.L. Role of Plk2 (Snk) in mouse development and cell proliferation. Mol. Cell. Biol. 2003, 23, 6936–6943. [Google Scholar] [CrossRef] [PubMed]
- Ou, B.; Zhao, J.; Guan, S.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Zheng, M.; Feng, H.; et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. 2016, 380, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Liu, Y.; Li, Q.; Chen, J. Polo-like kinase 2 promotes chemoresistance and predicts limited survival benefit from adjuvant chemotherapy in colorectal cancer. Int. J. Oncol. 2018, 52, 1401–1414. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wang, P.Y.; Ma, W.; Sung, H.J.; Matoba, S.; Hwang, P.M. Polo-like kinases mediate cell survival in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 14542–14546. [Google Scholar] [CrossRef]
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Song, T.S.; Huang, C.; Xu, J.R. Silencing of polo-like kinase 2 increases cell proliferation and decreases apoptosis in SGC-7901 gastric cancer cells. Mol. Med. Rep. 2015, 11, 3033–3038. [Google Scholar] [CrossRef]
- Syed, N.; Smith, P.; Sullivan, A.; Spender, L.C.; Dyer, M.; Karran, L.; O’Nions, J.; Allday, M.; Hoffmann, I.; Crawford, D.; et al. Transcriptional silencing of Polo-like kinase 2 (SNK/PLK2) is a frequent event in B-cell malignancies. Blood 2006, 107, 250–256. [Google Scholar] [CrossRef]
- Villegas, E.; Kabotyanski, E.B.; Shore, A.N.; Creighton, C.J.; Westbrook, T.F.; Rosen, J.M. Plk2 regulates mitotic spindle orientation and mammary gland development. Development 2014, 141, 1562–1571. [Google Scholar] [CrossRef]
- Matthew, E.M.; Yang, Z.; Peri, S.; Andrake, M.; Dunbrack, R.; Ross, E.; El-Deiry, W.S. Plk2 loss commonly occurs in colorectal carcinomas but not adenomas: Relationship to mTOR signaling. Neoplasia 2018, 20, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, S.; Zhao, Z.; Mao, X.; Huang, J.; Wu, Z.; Zheng, L.; Wang, Q. MicroRNA-27b up-regulated by human papillomavirus 16 E7 promotes proliferation and suppresses apoptosis by targeting polo-like kinase2 in cervical cancer. Oncotarget 2016, 7, 19666–19679. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Hou, N.; Ni, L.; Wang, A.Y.; Song, T.S.; et al. Mir-126 inhibits growth of SGC-7901 cells by synergistically targeting the oncogenes PI3KR2 and Crk, and the tumor suppressor PLK2. Int. J. Oncol. 2014, 45, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zha, Z.; Wang, H. Upregulation of microRNA-27a inhibits synovial angiogenesis and chondrocyte apoptosis in knee osteoarthritis rats through the inhibition of PLK2. J. Cell. Physiol. 2019, 234, 22972–22984. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Haupt, Y. Mutant p53 subverts PLK2 function in a novel, reinforced loop of corruption. Cell Cycle 2012, 11, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Akeno, N.; Miller, A.L.; Ma, X. Wikenheiser-Brokamp KA: p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene 2015, 34, 589–599. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.O.; Lee, S.K.; Kim, N.; You, G.Y.; Moon, J.W.; Sha, J.; Kim, S.J.; Park, S.H.; Kim, H.S. Celastrol suppresses breast cancer MCF-7 cell viability via the AMP-activated protein kinase (AMPK)-induced p53-polo like kinase 2 (PLK-2) pathway. Cell. Signal. 2013, 25, 805–813. [Google Scholar] [CrossRef]
- Ward, A.; Hudson, J.W. p53-Dependent and cell specific epigenetic regulation of the polo-like kinases under oxidative stress. PLoS ONE 2014, 9, e87918. [Google Scholar] [CrossRef]
- Li, J.; Ma, W.; Wang, P.; Hurley, P.J.; Bunz, F.; Hwang, P.M. Polo-like kinase 2 activates an antioxidant pathway to promote the survival of cells with mitochondrial dysfunction. Free Radic. Biol. Med. 2014, 73, 270–277. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange Consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 54, D1100–D1106. [Google Scholar] [CrossRef]
- Zou, H.H.; Yang, P.P.; Huang, T.L.; Zheng, X.X.; Xu, G.S. PLK2 plays an essential role in high D-glucose-induced apoptosis, ROS generation and inflammation in podocytes. Sci. Rep. 2017, 7, 4261. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Seal, B.S. Identification of a truncated nucleoprotein in avian metapneumovirus-infected cells encoded by a second AUG, in-frame to the full-length gene. Virol. J. 2005, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Paudel, S.; Shin, H. Role of trypsin in the replication of Avian metapneumovirus subtype C (strain MN-2a) and its entry into the Vero cells. Mol. Cell. Probes. 2015, 29, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Clubbe, J.; Naylor, C.J. Avian metapneumovirus M2:2 protein inhibits replication in Vero cells: Modification facilitates live vaccine development. Vaccine 2011, 29, 9493–9498. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.L.; Guan, X.L.; Liu, Y.Z.; Zhang, Y.; Wang, Y.Q.; Qi, X.L.; Cui, H.Y.; Liu, C.J.; Zhang, Y.P.; Gao, H.L.; et al. Integrin αvβ1 modulation affects subtype B avian metapneumovirus fusion protein-mediated cell-cell fusion and virus infection. J. Biol. Chem. 2016, 291, 14815–14825. [Google Scholar] [CrossRef]
- Munir, S.; Kaur, K.; Kapur, V. Avian metapneumovirus phosphoprotein targeted RNA interference silences the expression of viral proteins and inhibits virus replication. Antivir. Res. 2006, 69, 46–51. [Google Scholar] [CrossRef]
- Laconi, A.; Catelli, E.; Cecchinato, M.; Naylora, C.J. Two similar commercial live attenuated AMPV vaccines prepared by random passage of the identical field isolate, have unrelated sequences. Vaccine 2019, 37, 2765–2767. [Google Scholar] [CrossRef]
- de Graaf, M.; Schrauwen, E.J.A.; Herfst, S.; van Amerongen, G.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Fusion protein is the main determinant of metapneumovirus host tropism. J. Gen. Virol. 2009, 90, 1408–1416. [Google Scholar] [CrossRef]
- Nascimento, R.; Dias, J.D.; Parkhouse, R.M. The conserved UL24 family of human alpha, beta and gamma herpesviruses induces cell cycle arrest and inactivation of the cyclinB/cdc2 complex. Arch. Virol. 2009, 154, 1143–1149. [Google Scholar] [CrossRef]
- Vogt, A.; Scull, M.A.; Friling, T.; Horwitz, J.A.; Donovan, B.M.; Dorner, M.; Gerold, G.; Labitt, R.N.; Rice, C.M.; Ploss, A. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 2013, 444, 1–11. [Google Scholar] [CrossRef]
- Yuan, X.; Yao, Z.; Wu, J.; Zhou, Y.; Shan, Y.; Dong, B.; Zhao, Z.; Hua, P.; Chen, J.; Cong, Y. G1 phase cell cycle arrest induced by SARS-CoV 3a protein via the cyclin D3/pRb pathway. Am. J. Respir. Cell Mol. Biol. 2007, 37, 9–19. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, K.; Keiner, B.; Zhou, J.; Czudai, V.; Li, T.; Chen, Z.; Liu, J.; Klenk, H.D.; Shu, Y.L.; et al. Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2010, 84, 12832–12840. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Munday, D.C.; Howell, G.; Platt, G.; Barr, J.N.; Hiscox, J.A. Characterization of the interaction between human respiratory syncytial virus and the cell cycle in continuous cell culture and primary human airway epithelial cells. J. Virol. 2011, 85, 10300–10309. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Brown, C.M.; Westphal, D.; Ward, J.M.; Ward, V.K. Murine norovirus replication induces G0/G1 cell cycle arrest in asynchronously growing cells. J. Virol. 2015, 89, 6057–6066. [Google Scholar] [CrossRef] [PubMed]
- Quan, R.; Wei, L.; Zhu, S.; Wang, J.; Cao, Y.; Xue, C.; Yan, X.; Liu, J. Involvement of miR-15a in G0/G1 phase cell cycle arrest induced by porcine circovirus type 2 replication. Sci. Rep. 2016, 6, 27917. [Google Scholar] [CrossRef]
- Groskreutz, D.J.; Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Quelle, D.E.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus decreases p53 protein to prolong survival of airway epithelial cells. J. Immunol. 2007, 179, 2741–2747. [Google Scholar] [CrossRef]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef]
- Multhoff, G. Heat shock proteins in immunity. Handb. Exp. Pharmacol. 2006, 279–304. [Google Scholar]
- Everett, H.; McFadden, G. Apoptosis: An innate immune response to virus infection. Trends Microbiol. 1999, 7, 160–165. [Google Scholar] [CrossRef]
- van Hemert, M.J.; Steensma, H.Y.; van Heusden, G.P. 14-3-3 proteins: Key regulators of cell division, signalling and apoptosis. BioEssays 2001, 23, 936–946. [Google Scholar] [CrossRef]
- Aoki, H.; Hayashi, J.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J. Virol. 2000, 74, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.L.; Barnitz, R.A.; Sakai, K.; Lenardo, M.J. 14-3-3 theta binding to cell cycle regulatory factors is enhanced by HIV-1 Vpr. Biol. Direct 2008, 3, 17. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Accession No. | Ratios (Infection/Control) | Peptides | Sequence Coverage | |
|---|---|---|---|---|---|
| 48 hpi | 72 hpi | ||||
| 14-3-3 protein beta/alpha | F6XFG9 | 0.726 # | 0.792 | 16 | 70.33 |
| Cyclin-dependent kinase inhibitor 1B | F6Z4R0 | 0.732 # | 0.922 | 2 | 10.61 |
| Somatomedin-B | VTNC | 1.418 | 1.432 | 2 | 4.8 |
| 14-3-3 protein eta | F6RIX6 | 0.700# | 0.799 | 17 | 72.16 |
| 14-3-3 protein gamma | F6XLR8 | 0.689# | 0.778 | 18 | 85.02 |
| GTP-binding protein Rheb | F7H5C5 | 0.694# | 0.751 | 5 | 27.72 |
| Cyclin-dependent kinase 6 | F7E5V8 | 0.753# | 0.791 | 12 | 42.94 |
| Heat shock protein beta-1 | F6XTF7 | 0.759# | 0.818 | 16 | 84.88 |
| Non-specific serine/threonine protein kinase | F7GWI2 | 0.766# | 0.765# | 17 | 43.51 |
| Signal transducer and activator of transcription | F6WE02 | 0.651# | 0.732# | 24 | 38.67 |
| Cyclin-dependent kinase 1 isoform 1 | F7GRY6 | 0.745# | 0.738# | 13 | 53.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quan, R.; Wei, L.; Hou, L.; Wang, J.; Zhu, S.; Li, Z.; Lv, M.; Liu, J. Proteome Analysis in a Mammalian Cell Line Reveals that PLK2 Is Involved in Avian Metapneumovirus Type C (aMPV/C)-Induced Apoptosis. Viruses 2020, 12, 375. https://doi.org/10.3390/v12040375
Quan R, Wei L, Hou L, Wang J, Zhu S, Li Z, Lv M, Liu J. Proteome Analysis in a Mammalian Cell Line Reveals that PLK2 Is Involved in Avian Metapneumovirus Type C (aMPV/C)-Induced Apoptosis. Viruses. 2020; 12(4):375. https://doi.org/10.3390/v12040375
Chicago/Turabian StyleQuan, Rong, Li Wei, Lei Hou, Jing Wang, Shanshan Zhu, Zixuan Li, Moran Lv, and Jue Liu. 2020. "Proteome Analysis in a Mammalian Cell Line Reveals that PLK2 Is Involved in Avian Metapneumovirus Type C (aMPV/C)-Induced Apoptosis" Viruses 12, no. 4: 375. https://doi.org/10.3390/v12040375
APA StyleQuan, R., Wei, L., Hou, L., Wang, J., Zhu, S., Li, Z., Lv, M., & Liu, J. (2020). Proteome Analysis in a Mammalian Cell Line Reveals that PLK2 Is Involved in Avian Metapneumovirus Type C (aMPV/C)-Induced Apoptosis. Viruses, 12(4), 375. https://doi.org/10.3390/v12040375