Complete Genome Sequencing, Molecular Epidemiological, and Pathogenicity Analysis of Pigeon Paramyxoviruses Type 1 Isolated in Guangxi, China during 2012–2018

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Virus Isolation and Identification
2.3. Determination of Minimal Lethal Dose, Mean Death Time, and Intracerebral Pathogenicity Index
2.4. Cross-Reactivity Testing
2.5. RNA Extraction, Next-Generation Sequencing (NGS) Library Preparation, Sequencing, and Genome Assembly
2.6. Collection of Sequences and Evolutionary and Phylogenetic Analysis
2.7. Antigenicity and Hydrophilicity Analysis of F and HN Proteins
2.8. GenBank Accession Numbers
3. Results
3.1. Virus Isolation and In Vivo Pathogenicity Characterization
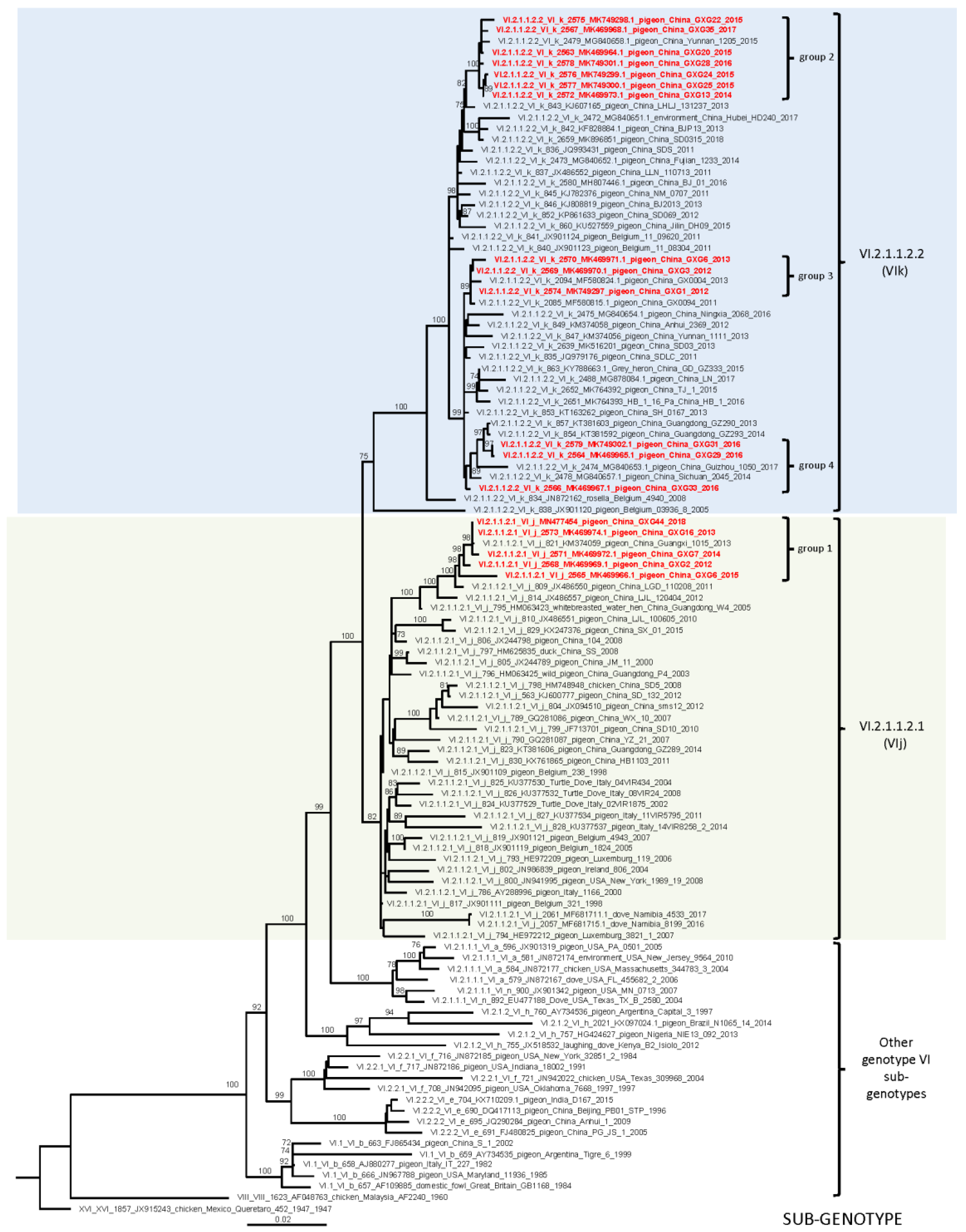
3.2. Whole Genome Sequencing and Phylogenetic Analyses
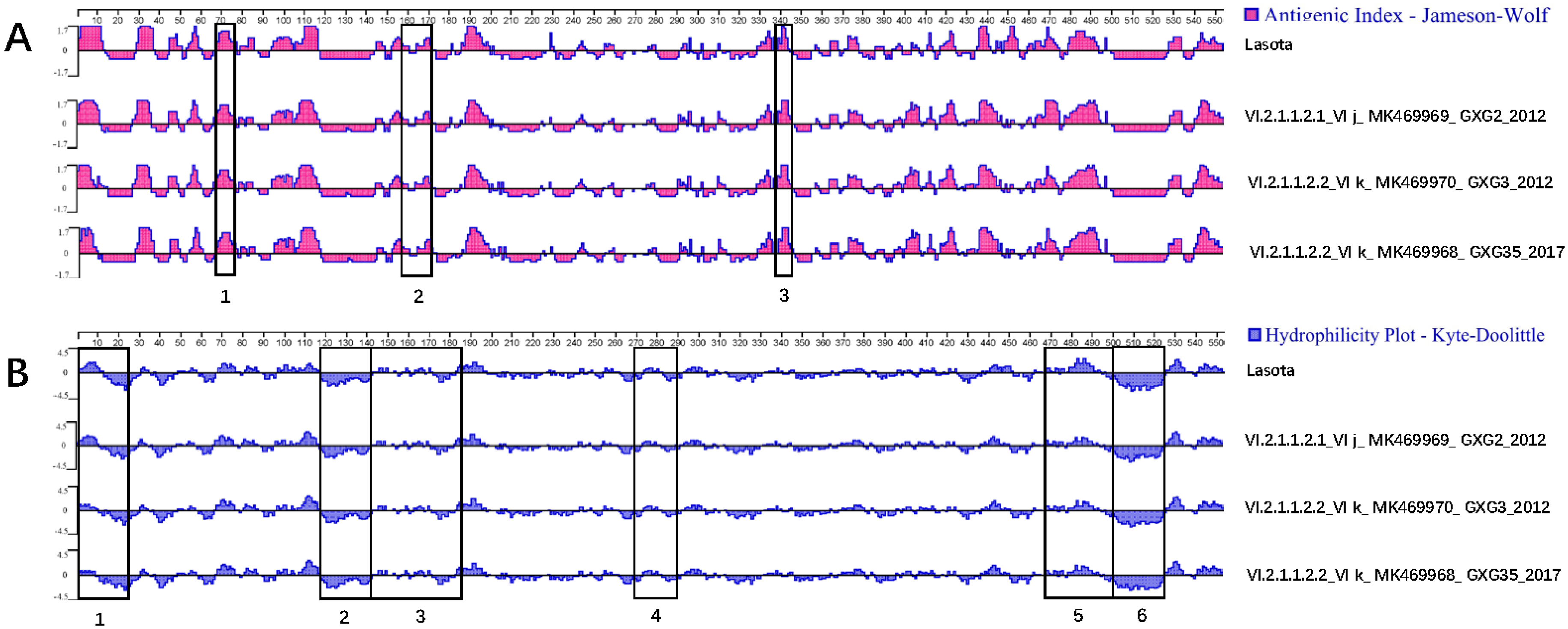
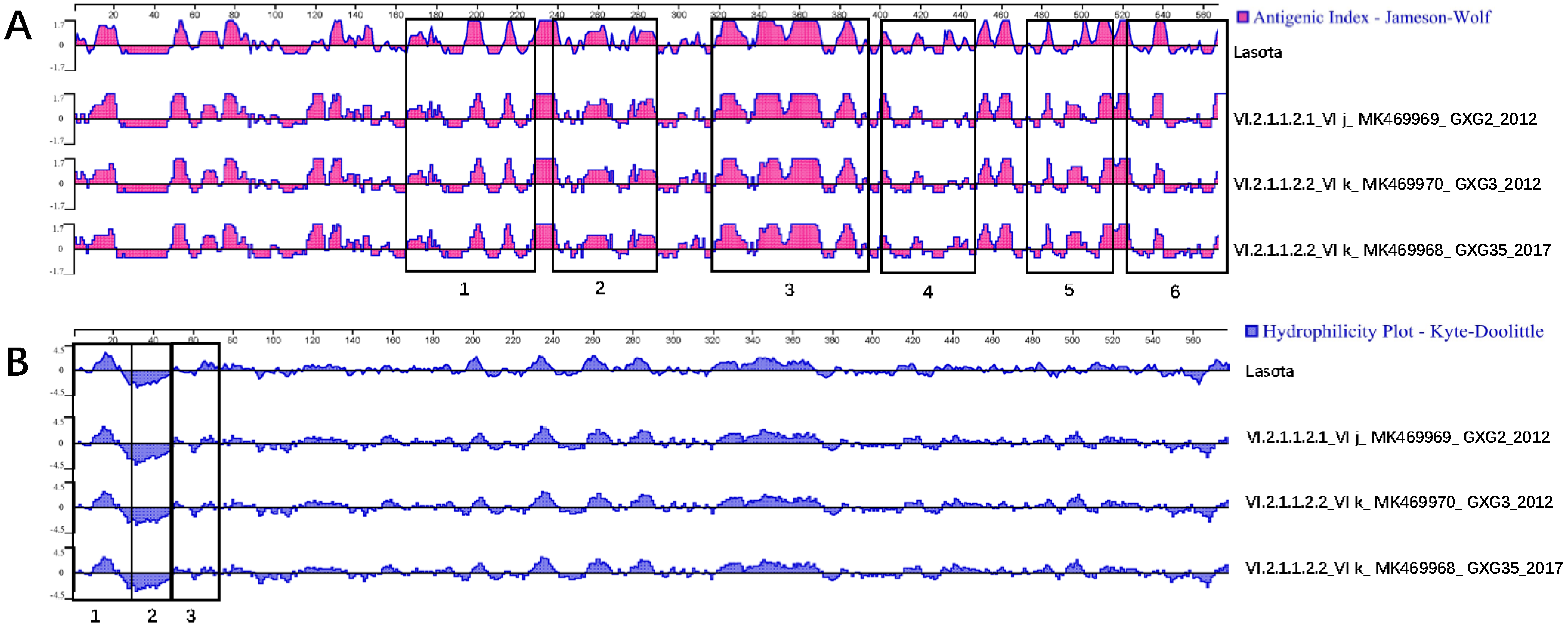
3.3. Serum Cross-Reactivity, Antigenicity, and Hydrophilicity Testing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amarasinghe, G.K.; Ayllon, M.A.; Bao, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.J. Newcastle disease and other avian paramyxoviruses. Rev. Sci Tech. 2000, 19, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, K.M.; Abolnik, C.; Afonso, C.L.; Albina, E.; Bahl, J.; Berg, M.; Briand, F.X.; Brown, I.H.; Choi, K.S.; Chvala, I.; et al. Updated unified phylogenetic classification system and revised nomenclature for Newcastle disease virus. Infect. Genet. Evol. 2019, 74, 103917. [Google Scholar] [CrossRef] [PubMed]
- Molini, U.; Aikukutu, G.; Khaiseb, S.; Cattoli, G.; Dundon, W.G. Phylogenetic Analysis of Pigeon Paramyxoviruses Type-1 Identified in Mourning Collared-doves (Streptopelia decipiens) in Namibia, Africa. J. Wildl. Dis. 2018, 54, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.S.; Alexander, D.J.; Brockman, S.; Kemp, P.A.; Manvell, R.J. Evaluation of mouse monoclonal antibodies raised against an isolate of the variant avian paramyxovirus type 1 responsible for the current panzootic in pigeons. Arch. Virol. 1989, 104, 53–61. [Google Scholar] [CrossRef]
- de Leeuw, O.; Peeters, B. Complete nucleotide sequence of Newcastle disease virus: Evidence for the existence of a new genus within the subfamily Paramyxovirinae. J. Gen. Virol. 1999, 80, 131–136. [Google Scholar] [CrossRef]
- Hanson, R.P.; Brandly, C.A. Identification of vaccine strains of Newcastle disease virus. Science 1955, 122, 156–157. [Google Scholar]
- Alexander, D.J. Newcastle disease and other avian Paramyxoviridae infections. In Diseases of Poultry, 10th ed.; Calnek, B.W., Barnes, H.J., Beard, C.W., McDougald, L.R., Saif, Y.M., Eds.; Iowa State University Press: Ames, IA, USA, 1997; pp. 541–569. [Google Scholar]
- Kim, L.M.; King, D.J.; Guzman, H.; Tesh, R.B.; Rosa, A.P.A.T.D.; Bueno, R.; Dennett, J.A.; Afonso, C.L. Biological and phylogenetic characterization of pigeon paramyxovirus serotype 1 circulating in wild North American pigeons and doves. J. Clin. Microbiol. 2008, 46, 3303–3310. [Google Scholar] [CrossRef]
- Miller, P.J.; Haddas, R.; Simanov, L.; Lublin, A.; Rehmani, S.F.; Wajid, A.; Bibi, T.; Khan, T.A.; Yaqub, T.; Setiyaningsih, S.; et al. Identification of new sub-genotypes of virulent Newcastle disease virus with potential panzootic features. Infect. Genet. Evol. 2015, 29, 216–229. [Google Scholar] [CrossRef]
- Kaleta, E.F.; Alexander, D.J.; Russell, P.H. The first isolation of the avian PMV-1 virus responsible for the current panzootic in pigeons? Avian Pathol. 1985, 14, 553–557. [Google Scholar] [CrossRef]
- Wang, J.; Liu, H.; Liu, W.; Zheng, D.; Zhao, Y.; Li, Y.; Wang, Y.; Ge, S.; Lv, Y.; Zuo, Y.; et al. Genomic Characterizations of Six Pigeon Paramyxovirus Type 1 Viruses Isolated from Live Bird Markets in China during 2011 to 2013. PLoS ONE 2015, 10, e0124261. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Qu, Y.; Wang, F.; Liu, S.; Sun, H. Genotypic and pathotypic characterization of Newcastle disease virus isolated from racing pigeons in China. Poult. Sci. 2015, 94, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Z.; Song, C.; Wang, Y.; Yu, B.; Zheng, D.; Sun, C.; Wu, Y. Characterization of Pigeon-Origin Newcastle Disease Virus Isolated in China. Avian Dis. 2006, 50, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Deng, Q.; Li, H.; Pan, C.; Zhai, G.; Yuan, Y.; Cheng, E.; Zhang, Y.; Mo, M.; Huang, T. Molecular characterization of two novel sub-sublineages of pigeon paramyxovirus type 1 in China. Arch. Virol. 2018, 163, 2971–2984. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Xu, X.; Yin, R.; Qian, J.; Sun, Y.; Wang, C.; Ding, C.; Yu, S.; Hu, S.; Liu, X.; et al. Identification and pathotypical analysis of a novel VIk sub-genotype Newcastle disease virus obtained from pigeon in China. Virus Res. 2017, 238, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.J.; Swayne, D.E. Newcastle disease virus and other avian paramyxoviruses. In A Laboratory Manual for the Isolation and Identification of Avian Pathogens; Swayne, D.E., Glisson, J.R., Jackwood, M.W., Pearson, J.E., Eds.; The American Association of Avian Pathologists: Kennett Square, PA, USA, 1998; pp. 156–163. [Google Scholar]
- Song, D.; Huang, D.; Peng, Q.; Huang, T.; Chen, Y.; Zhang, T.; Nie, X.; He, H.; Wang, P.; Liu, Q.; et al. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea viruses associated with outbreaks of severe diarrhea in piglets in Jiangxi, China 2013. PLoS ONE 2015, 10, e0120310. [Google Scholar] [CrossRef] [PubMed]
- OIE. Newcastle disease. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals: Mammals, Birds and Bees; Biological Standards Commission, World Organization for Animal Health: Paris, France, 2012; pp. 555–574. [Google Scholar]
- Qiu, X.; Meng, C.; Zhan, Y.; Yu, S.; Li, S.; Ren, T.; Yuan, W.; Xu, S.; Sun, Y.; Tan, L.; et al. Phylogenetic, antigenic and biological characterization of pigeon paramyxovirus type 1 circulating in China. Virol. J. 2017, 14, 186. [Google Scholar] [CrossRef]
- Archetti, I.; Horsfall, F.L., Jr. Persistent antigenic variation of influenza A viruses after incomplete neutralization in ovo with heterologous immune serum. J. Exp. Med. 1950, 92, 441–462. [Google Scholar] [CrossRef]
- Yang, H.M.; Zhao, J.; Xue, J.; Yang, Y.L.; Zhang, G.Z. Antigenic variation of LaSota and genotype VII Newcastle disease virus (NDV) and their efficacy against challenge with velogenic NDV. Vaccine 2017, 35, 27–32. [Google Scholar] [CrossRef]
- He, Y.; Taylor, T.L.; Dimitrov, K.M.; Butt, S.L.; Stanton, J.B.; Goraichuk, I.V.; Fenton, H.; Poulson, R.; Zhang, J.; Brown, C.C. Whole-genome sequencing of genotype VI Newcastle disease viruses from formalin-fixed paraffin-embedded tissues from wild pigeons reveals continuous evolution and previously unrecognized genetic diversity in the US. Virol. J. 2018, 15, 9. [Google Scholar] [CrossRef]
- Sabra, M.; Dimitrov, K.M.; Goraichuk, I.V.; Wajid, A.; Sharma, P.; Williams-Coplin, D.; Basharat, A.; Rehmani, S.F.; Muzyka, D.V.; Miller, P.J.; et al. Phylogenetic assessment reveals continuous evolution and circulation of pigeon-derived virulent avian avulaviruses 1 in Eastern Europe, Asia, and Africa. BMC Vet. Res. 2017, 13, 291. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, K.M.; Sharma, P.; Volkening, J.D.; Goraichuk, I.V.; Wajid, A.; Rehmani, S.F.; Basharat, A.; Shittu, I.; Joannis, T.M.; Miller, P.J.; et al. A robust and cost-effective approach to sequence and analyze complete genomes of small RNA viruses. Virol. J. 2017, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 45, D37–D42. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. In Lectures on Mathematics in the Life Sciences; Miura, R.M., Ed.; American Mathematical Society: Providence, RI, USA, 1986; pp. 57–86. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Venugopal, N.; Fadly, A. Diseases of Poultry, 13th ed.; Swayne, D.E., Ed.; Wiley-Blackwell: Ames, IA, USA, 2013. [Google Scholar]
- Dortmans, J.C.; Fuller, C.M.; Aldous, E.W.; Rottier, P.J.; Peeters, B.P. Two genetically closely related pigeon paramyxovirus type 1 (PPMV-1) variants with identical velogenic fusion protein cleavage sites but with strongly contrasting virulence. Vet. Microbiol. 2010, 143, 139–144. [Google Scholar] [CrossRef]
- Dortmans, J.C.; Koch, G.; Rottier, P.J.; Peeters, B.P. Virulence of pigeon paramyxovirus type 1 does not always correlate with the cleavability of its fusion protein. J. Gen. Virol. 2009, 90, 2746–2750. [Google Scholar] [CrossRef]
- Dortmans, J.; Rottier, P.; Koch, G.; Peeters, B. Passaging of a Newcastle disease virus pigeon variant in chickens results in selection of viruses with mutations in the polymerase complex enhancing virus replication and virulence. J. Gen. Virol. 2011, 92, 336–345. [Google Scholar] [CrossRef]
- Ferreira, H.L.; Taylor, T.L.; Dimitrov, K.M.; Sabra, M.; Afonso, C.L.; Suarez, D.L. Virulent Newcastle disease viruses from chicken origin are more pathogenic and transmissible to chickens than viruses normally maintained in wild birds. Vet. Microbiol. 2019, 235, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, X.; Xu, Y.; Han, Z.; Shao, Y.; Kong, X.; Liu, S. A comparative study of pigeons and chickens experimentally infected with PPMV-1 to determine antigenic relationships between PPMV-1 and NDV strains. Vet. Microbiol. 2014, 168, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Abolnik, C.; Gerdes, G.H.; Kitching, J.; Swanepoel, S.; Romito, M.; Bisschop, S.P. Characterization of pigeon paramyxoviruses (Newcastle disease virus) isolated in South Africa from 2001 to 2006. Onderstepoort J. Vet. Res. 2008, 75, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Meulemans, G.; Berg, T.v.d.; Decaesstecker, M.; Boschmans, M. Evolution of pigeon Newcastle disease virus strains. Avian Pathol. 2002, 31, 515–519. [Google Scholar]
- Moura, V.M.; Susta, L.; Cardenas-Garcia, S.; Stanton, J.B.; Miller, P.J.; Afonso, C.L.; Brown, C.C. Neuropathogenic Capacity of Lentogenic, Mesogenic, and Velogenic Newcastle Disease Virus Strains in Day-Old Chickens. Vet. Pathol. 2016, 53, 53–64. [Google Scholar] [CrossRef]
- Kommers, G.D.; King, D.J.; Seal, B.S.; Brown, C.C. Virulence of pigeon-origin Newcastle disease virus isolates for domestic chickens. Avian Dis. 2001, 45, 906–921. [Google Scholar]
- Dimitrov, K.M.; Ramey, A.M.; Qiu, X.; Bahl, J.; Afonso, C.L. Temporal, geographic, and host distribution of avian paramyxovirus 1 (Newcastle disease virus). Infect. Genet. Evol. 2016, 39, 22–34. [Google Scholar] [CrossRef]
- Teske, L.; Ryll, M.; Rautenschlein, S. Epidemiological investigations on the role of clinically healthy racing pigeons as a reservoir for avian paramyxovirus-1 and avian influenza virus. Avian Pathol. 2013, 42, 557–565. [Google Scholar] [CrossRef]
- Alexander, D.J.; Aldous, E.W.; Fuller, C.M. The long view: A selective review of 40 years of Newcastle disease research. Avian Pathol. 2012, 41, 329–335. [Google Scholar] [CrossRef]
- Aldous, E.W.; Fuller, C.M.; Ridgeon, J.H.; Irvine, R.M.; Alexander, D.J.; Brown, I.H. The evolution of pigeon paramyxovirus type 1 (PPMV-1) in Great Britain: A molecular epidemiological study. Transbound. Emerg. Dis. 2014, 61, 134–139. [Google Scholar] [CrossRef]
- Heath, T.A.; Hedtke, S.M.; Hillis, D.M. Taxon sampling and the accuracy of phylogenetic analyses. J. Syst. Evol. 2008, 46, 239–257. [Google Scholar] [CrossRef]
- Wang, J.Y.; Liu, W.H.; Ren, J.J.; Tang, P.; Wu, N.; Wu, H.Y.; Ching, C.D.; Liu, H.J. Characterization of emerging Newcastle disease virus isolates in China. Virol. J. 2015, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Terregino, C.; Aldous, E.W.; Heidari, A.; Fuller, C.M.; De Nardi, R.; Manvell, R.J.; Beato, M.S.; Shell, W.M.; Monne, I.; Brown, I.H.; et al. Antigenic and genetic analyses of isolate APMV/wigeon/Italy/3920-1/2005 indicate that it represents a new avian paramyxovirus (APMV-12). Arch. Virol. 2013, 158, 2233–2243. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Isolates | GenBank Accession No. | Isolation Date | Flock Size | Clinical Signs and Gross Lesions | HA Titer | MDT | ICPI | NGS Raw Reads | Filtered Reads | PPMV-1 Reads | Genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GXG1 | MK749297 | Nov 2012 | 800 | neurological symptoms, diarrhea | log28 | 76 h | 0.00 | 15,033,538 | 14,121,738 | 7,788,836 | VI.2.1.1.2.2 (VIk) |
| GXG2 | MK469969 | Mar 2012 | 4000 | neurological symptoms, hemorrhages in glandular stomach | log28 | 94 h | 0.00 | 14,119,609 | 13,203,568 | 1,630,974 | VI.2.1.1.2.1 (VIj) |
| GXG3 | MK469970 | May 2012 | 1000 | hemorrhages in glandular stomach and peritonitis | log29 | 72 h | 0.25 | 13,767,808 | 12,781,443 | 1,755,997 | VI.2.1.1.2.2 (VIk) |
| GXG6 | MK469971 | May 2013 | 2500 | diarrhea, hemorrhages, and necrosis in the spleen | log27 | 92 h | 0.00 | 14,546,579 | 13,527,020 | 2,989,862 | VI.2.1.1.2.2 (Vik) |
| GXG7 | MK469972 | Feb 2014 | 3600 | neurological symptoms, diarrhea | log210 | 81 h | 0.25 | 15,209,534 | 14,368,697 | 4,024,270 | VI.2.1.1.2.1 (VIj) |
| GXG13 | MK469973 | Mar 2015 | 2000 | neurological symptoms | log27 | 62 h | 0.17 | 14,891,909 | 13,844,914 | 3,051,814 | VI.2.1.1.2.2 (VIk) |
| GXG16 | MK469974 | May 2015 | 2000 | neurological symptoms, diarrhea | log24 | 69 h | 0.63 | 13,082,370 | 12,098,553 | 1,630,974 | VI.2.1.1.2.1 (VIj) |
| GXG20 | MK469964 | Jun 2015 | 1000 | neurological symptoms | log27 | 78 h | 0.00 | 10,876,496 | 9,871,185 | 2,172,462 | VI.2.1.1.2.2 (VIk) |
| GXG22 | MK749298 | Jul 2015 | 400 | hemorrhages in glandular stomach, neurologic symptoms | log25 | 80 h | 0.00 | 12,076,535 | 11,249,343 | 1,306,032 | VI.2.1.1.2.2 (VIk) |
| GXG24 | MK749299 | Jul 2015 | 1200 | diarrhea, hemorrhages, and necrosis in the spleen | log23 | 105 h | 0.00 | 13,863,796 | 12,818,138 | 1,489,872 | VI.2.1.1.2.2 (VIk) |
| GXG25 | MK749300 | Aug 2015 | 1200 | hemorrhages in glandular stomach, neurologic symptoms | log23 | 96 h | 0.00 | 14,886,898 | 13,994,957 | 4,915,176 | VI.2.1.1.2.2 (VIk) |
| GXG6 | MK469966 | Sep 2015 | 2200 | neurological symptoms, diarrhea | log23 | 114 h | 0.00 | 10,669,774 | 10,033,373 | 2,887,488 | VI.2.1.1.2.1 (VIj) |
| GXG28 | MK749301 | Jan 2016 | 1000 | diarrhea, hemorrhages in glandular stomach | log24 | 90 h | 0.00 | 17,058,483 | 15,953,502 | 7,449,482 | VI.2.1.1.2.2 (VIk) |
| GXG29 | MK469965 | Feb 2016 | 4000 | neurological symptoms | log25 | 70 h | 0.00 | 14,457,740 | 13,485,935 | 7,797,452 | VI.2.1.1.2.2 (VIk) |
| GXG31 | MK749302 | Mar 2016 | 4000 | neurological symptoms | log23 | 93 h | 0.00 | 13,384,970 | 12,544,326 | 2,102,690 | VI.2.1.1.2.2 (VIk) |
| GXG33 | MK469967 | Apr 2016 | 1200 | neurological symptoms | log26 | 69 h | 0.45 | 9,915,567 | 9,426,738 | 1,350,934 | VI.2.1.1.2.2 (VIk) |
| GXG35 | MK469968 | Jun 2017 | 2600 | hemorrhages in glandular and stomach | log23 | 98 h | 0.00 | 12,150,471 | 10,993,904 | 1,713,840 | VI.2.1.1.2.2 (VIk) |
| GXG44 | MN477454 | Dec 2018 | 3000 | neurologic symptoms, diarrhea | log24 | 96 h | 0.00 | 15,825,591 | 14,138,234 | 4,824,912 | VI.2.1.1.2.2 (VIj) |
| Number of Base Substitutions per Site a | ||||||
|---|---|---|---|---|---|---|
| VI.1 | VI.2.2.1 | VI.2.2.2 | VI.2.1.1.1 | VI.2.1.2 | VI.2.1.1.2.1 | |
| VI.1 | ||||||
| VI.2.2.1 | 6.5 | |||||
| VI.2.2.2 | 7.2 | 6.2 | ||||
| VI.2.1.1.1 | 8.5 | 8.3 | 9.0 | |||
| VI.2.1.2 | 8.2 | 8.9 | 9.3 | 7.9 | ||
| VI.2.1.1.2.1 | 7.8 | 7.6 | 8.4 | 6.3 | 7.8 | |
| VI.2.1.1.2.2 | 8.4 | 8.5 | 9.3 | 7.2 | 8.9 | 5.3 |
| Ab/Ag | Lasota | GXG2 | GXG7 | GXG16 | GXG6/2015 | GXG1 | GXG3 | GXG6/2013 | GXG13 | GXG24 |
|---|---|---|---|---|---|---|---|---|---|---|
| Lasota | ||||||||||
| GXG2 | 0.75 | |||||||||
| GXG7 | 0.77 | 0.91 | ||||||||
| GXG16 | 0.79 | 0.90 | 0.93 | |||||||
| GXG6/2015 | 0.75 | 0.94 | 0.89 | 0.95 | ||||||
| GXG1 | 0.66 | 0.92 | 0.92 | 0.85 | 0.88 | |||||
| GXG3 | 0.81 | 0.98 | 1.00 | 0.97 | 0.94 | 0.98 | ||||
| GXG6/2013 | 0.79 | 0.90 | 0.91 | 0.88 | 0.85 | 0.85 | 0.94 | |||
| GXG13 | 0.71 | 0.91 | 0.89 | 0.91 | 0.85 | 0.89 | 1.00 | 0.92 | ||
| GXG24 | 0.78 | 0.90 | 0.85 | 0.90 | 0.85 | 0.85 | 0.92 | 0.88 | 0.87 | |
| GXG25 | 0.68 | 0.93 | 0.89 | 0.88 | 0.87 | 0.92 | 0.97 | 0.91 | 0.93 | 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Lu, B.; Dimitrov, K.M.; Liang, J.; Chen, Z.; Zhao, W.; Qin, Y.; Duan, Q.; Zhou, Y.; Liu, L.; et al. Complete Genome Sequencing, Molecular Epidemiological, and Pathogenicity Analysis of Pigeon Paramyxoviruses Type 1 Isolated in Guangxi, China during 2012–2018. Viruses 2020, 12, 366. https://doi.org/10.3390/v12040366
He Y, Lu B, Dimitrov KM, Liang J, Chen Z, Zhao W, Qin Y, Duan Q, Zhou Y, Liu L, et al. Complete Genome Sequencing, Molecular Epidemiological, and Pathogenicity Analysis of Pigeon Paramyxoviruses Type 1 Isolated in Guangxi, China during 2012–2018. Viruses. 2020; 12(4):366. https://doi.org/10.3390/v12040366
Chicago/Turabian StyleHe, Ying, Bingxia Lu, Kiril M. Dimitrov, Jiaxing Liang, Zhongwei Chen, Wu Zhao, Yibin Qin, Qunpeng Duan, Yingning Zhou, Lei Liu, and et al. 2020. "Complete Genome Sequencing, Molecular Epidemiological, and Pathogenicity Analysis of Pigeon Paramyxoviruses Type 1 Isolated in Guangxi, China during 2012–2018" Viruses 12, no. 4: 366. https://doi.org/10.3390/v12040366
APA StyleHe, Y., Lu, B., Dimitrov, K. M., Liang, J., Chen, Z., Zhao, W., Qin, Y., Duan, Q., Zhou, Y., Liu, L., Li, B., Yu, L., Duan, Z., & Liu, Q. (2020). Complete Genome Sequencing, Molecular Epidemiological, and Pathogenicity Analysis of Pigeon Paramyxoviruses Type 1 Isolated in Guangxi, China during 2012–2018. Viruses, 12(4), 366. https://doi.org/10.3390/v12040366