2. Materials and Methods
2.1. Materials and Experimental Instruments
Human cervical cancer cell lines CaSki and HeLa were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). SP2/0-Ag14 mouse myeloma cells were obtained from Shanghai Binsuirtez Biotechnology Co., Ltd. All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Waltham, MA, USA) plus 10% fetal bovine serum (FBS, Biological Industries, Kibbutz Beit-Haemek, Israel). Primers were synthesized by Sangon Biotech (Shanghai, China). Polyethylene Glycol 1500 (14292800, Roche, Basel, Switzerland), Freund’s Adjuvant, Complete (F881-6, SIGMA), Freund’s Adjuvant, Incomplete (F5506-10), Hypoxanthine-Aminopterin-Thymidine (HAT) MediaSupplement (50×) Hybri-MaxTM (H0262-10, SIGMA), Hypoxanthine and Thymidine (HT) MediaSupplement (50×) Hybri-MaxTM (H137-10, SIGMA), goat anti-mouse IgG-horseradish peroxidase (HRP) (Invitrogen), tetramethylbenzidine (TMB) chromogen solution for ELISA (PR1210, Solarbio, Beijing, China), a Peroxidase Labeling Kit-NH2 (LK51, Dojindo, Shanghai, China), a Biotin Labeling Kit-NH2 (LK55, Dojindo), and a Smart Electrochemiluminescence (ECL) Basic Luminol (Smart-Lifesciences, Changzhou, China) were obtained. Normal cervical tissues, HPV16/18-positive cervical cancer tissues, and paraffin sections were provided by The Ninth People’s Hospital of Chongqing. One paraffin tissue section from a patient with HPV16-positive cervical squamous cell invasive carcinoma in stage Ib and one paraffin tissue section from a patient with HPV18-positive cervical squamous adenocarcinoma in stage IIa were used in the immunohistochemistry (IHC) assay. Clinical specimens used in chemiluminescence immunoassay included 4 cases of normal cervical tissue masses derived from 4 patients with uterine fibroids, 4 cases of cervical cancer tissues derived from 4 patients with stage Ib HPV52-positive cervical squamous cell invasive carcinoma, 7 cases of cervical cancer tissues derived from patients with stage Ib HPV16-positive cervical squamous cell carcinoma, and 5 cases of cervical cancer tissues derived from stage IIa HPV16-positive cervical cancers. All cancerous specimens were obtained by using the extensive hysterectomy, while normal specimens were obtained by using the panhysterectomy. A Mini Shaker MH-1 microplate oscillator (Kylin-Bell Lab Instruments, Haimen, Jiangsu, China), a 1510 MULTISKAN GO full-wavelength microplate reader (Thermo Fisher Scientific, Waltham, MA, USA), and an Infinite® M200 PRO microplate reader (TECAN, Männedorf, Switzerland) were used for general experiments.
2.2. Preparation of HPV16 E7-HIS Fusion Oncoprotein
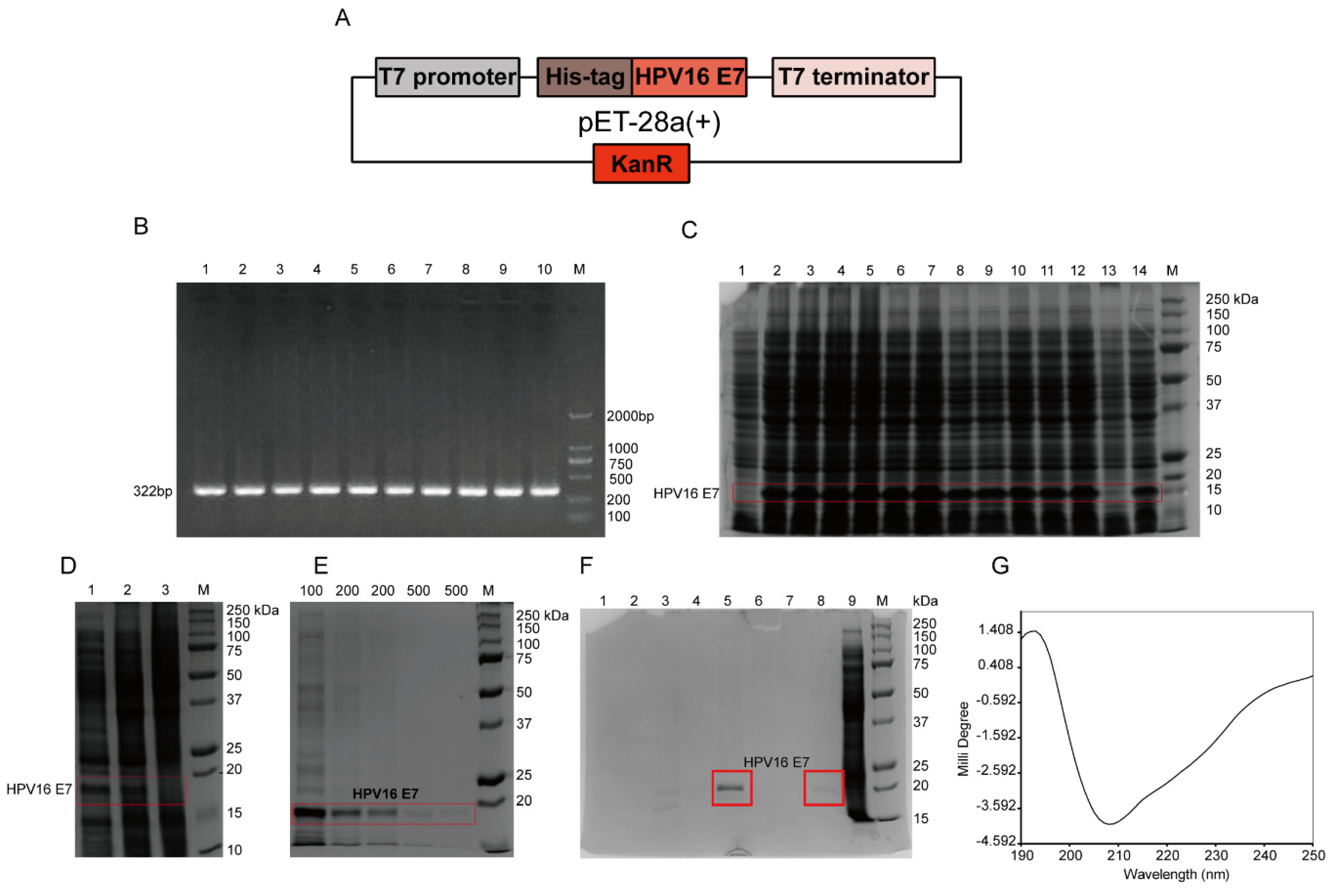
According to the full-length HPV16 E7 gene, specific primers with 6 histidine (HIS) tags were designed. The sequences of primers F and R were as follows: F: 5′-GCCCATGGACCATCACCATCACCATATGCATGGAGATACACCTAC-3′; R: 5′-GCAAGCTTTTATGGTTTCTGYGAACAGATGGGGCACAC-3′. Using the total genome DNA of CaSki cells, which was pre-extracted by using the Tissue/Cell DNA Extraction Kit (Shanghai Huashun Biothech Co. ltd., China), as a template, the full-length HPV16 E7 gene was amplified by PCR. The gradient PCR reaction conditions were performed as follows: 94 °C pre-denaturation for 5 min; 94 °C for 30 s; 10 annealing temperatures, 55.1, 55.5, 56.3, 57.7, 59.4, 61.4, 63.3, 65.3, 67.6, 69.0, 69.7, and 70.2 °C for 30 s, respectively; 35 cycles of 72 °C for 60 s; and 72 °C extension for 7 min. The 322 bp product included the full-length HPV16 E7 gene of 297 bp, plus 25 bp of the following bases: 2 protective bases, GC; NcoI enzyme restriction sites, CCATGG and 17 bases of histidine tag gene (ACCATCACCATCACCAT), which was without one base C after the frameshift. The amplified product was digested with enzymes and identified by sequencing, and the gene was inserted into the plasmid pET-28a(+) containing T7 promoter to construct an expression vector. Then it was transformed into competent cells Rosetta (DE3) pLysS, and the engineered bacteria pET-28a(+)-HPV16 E7-Rosetta (DE3) plysS was induced by 1 mM isopropyl-β-d-thiogalactoside (IPTG; Sangon, Shanghai, China) in Luria-Bertani (LB) medium at 37 °C for 4 h. Then the bacterial solution was centrifuged, and 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to determine the protein expression level and expression form. Nickel column affinity chromatography was used for the purification of the HPV16 E7-HIS recombinant oncoprotein. The secondary structure of the purified HPV16 E7-HIS fusion oncoprotein was determined by circular dichroism chromatography.
2.3. Preparation of Mouse anti-HPV16 E7-HIS Fusion Oncoprotein mAbs
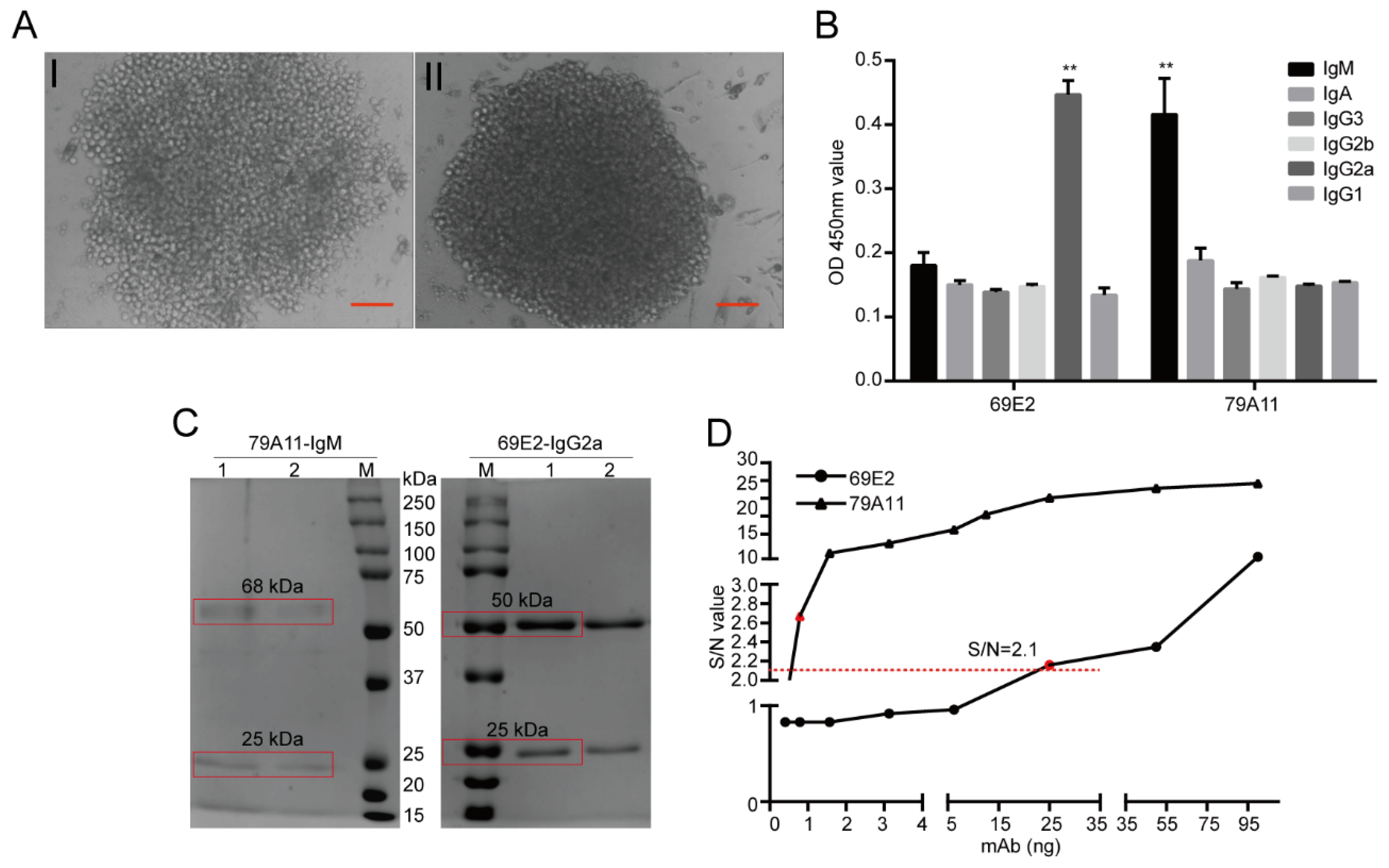
The HPV16 E7-HIS fusion oncoprotein was injected at a dose of 100 μg per BALB/c mouse through the footpad of the hind paw and the abdominal cavity. Footpad immunization was used to stimulate B lymphocyte proliferation and antibody production in the inguinal lymph nodes of the mice. Abdominal immunization was used to mainly stimulate mouse B lymphocyte proliferation and antibody production in the spleen. Inguinal lymph node cell suspension and spleen cell suspension of the same mouse, both of which are with high serum antibody titers, were respectively performed to fuse with SP2/0-Ag14 cells. The animals were raised in a specific pathogen free (SPF) room and the feeding conditions were strictly standardized. Before the antibodies were collected and the mice sacrificed, nasal anesthesia (isoflurane) was used to reduce their pain. The animal experiment was pre-approved by the Systems Biology and Institutional Animal Care and Use Committees of Southwest University (IACUC No.: 20180505-07; Approval date: 5 May 2018) and supervised by the Institutional Animal Care and Use Committees of the Institute of Sericulture. Monoclonal antibody subtypes were identified using mouse monoclonal antibody isotyping reagents (SIGMA). Ammonium sulfate precipitation, Protein G, Protein A, and IgM affinity chromatography, and gel chromatography were used to purify the mAb ascites, thereby obtaining several mouse mAbs for the HPV16 E7 oncoprotein with high purity.
2.4. Characterization of Mouse anti-HPV16 E7-HIS Fusion Oncoprotein mAbs by Using Immunocytochemistry (ICC), Immunofluorescence (IF), Immunohistochemistry (IHC), and Western Blot (WB)
CaSki and HeLa cell lines were cultured on the Fisherbrand microscope cover glasses (Cat. No. 125-45-83; Thermo Fisher) in 24-well plates with 20,000 cells per well. The mAbs were performed in the ICC and IF reactions. The SP2/0-Ag14 cell supernatant was used as a negative control. Additionally, a control without a primary antibody as a blank control was also set.
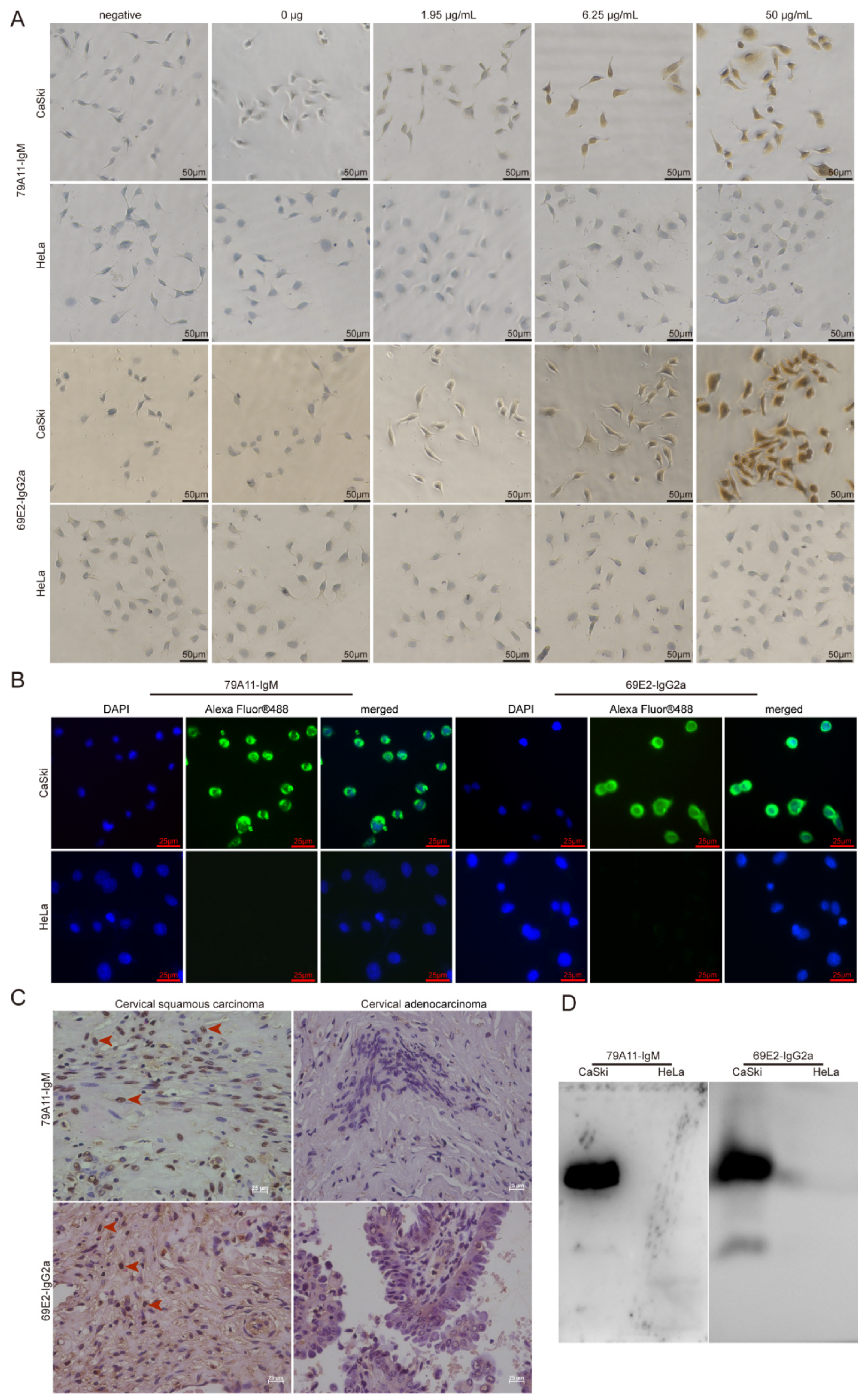
The detailed steps of the immunocytochemical reaction were as follows: CaSki and Hela cells cultured on the cover glasses in 24-well plates were fixed with 4% paraformaldehyde for 15 min at room temperature (RT). After washing, 3% H2O2 was used to eliminate endogenous peroxidase activity at RT for 20 min. Then 200 µL 1.95, 6.25, 50 µg/mL 79A11, and 69E2 primary antibodies were added to the corresponding wells, respectively. Fifty microliters of magnifying agent (Reagent I in the Dolink-2 Plus ® Polymer HRP Detection System from Zsbio Inc., Beijing, China, Cat. No. PV-9002) was added to each well, and incubated at 37 °C for 1 h. One hundred and fifty microliters of secondary antibody HRP-labeled anti-mouse IgG polymer (Reagent II in the Dolink-2 Plus ® Polymer HRP Detection System) with phosphate buffer saline (PBS) at 1:15,000 was added and incubated at 37 °C for 1 h. A diaminobenzadine (DAB; Cat. No.: ZLI-9018, ZsBio) color development solution was used for coloration. One hundred microliters of hematoxylin staining was used for 6–7 min for counterstaining. After being washed with water for 3 times, 100 µL 1% hydrochloric acid alcohol was used for differentiation. Then 0.2% ammonium hydroxide was used for bluing before washing with water under a microscope.
Preliminary application of mAbs 79A11 and 69E2 in indirect IF was performed as follows: CaSki and HeLa cells were cultured on the cover glasses. Ten micrograms/milliliter of primary antibodies (79A11 and 69E2) with PBS were added, and placed at 4 °C overnight for 18–20 h. Then the cells were incubated at RT for a while and then at 37 °C for 1.5 h. After washing with PBS containing 0.05% Tween 20 (PBST) 5 times, each time for 3 min, and 100 μL PBS-diluted fluorescein-labeled goat anti-mouse IgG at 1:1000, the sample was incubated at RT for 2 h. Then it was washed 5 times with PBST for 3 min each time. Afterwards, 100 µL 5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) staining solution (Beyotime, Shanghai, China) was added and the sample incubated at RT for 20–30 min, and then washed 5 times with PBST for 3 min each time. Then the cells were observed under a fluorescence microscope.
HPV16-positive cervical squamous cancer and HPV18-positive cervical cancer adenocarcinoma paraffin sections were used in IHC. Detailed protocols were as follows: paraffin specimen sections were heated at 60 °C for 2 h. Multiple steps were performed for dewaxing and hydration: xylene I for 15 min, xylene II for 15 min, 1/2 xylene (xylene:absolute ethanol = 1:1) for 10 min, absolute ethanol for 10 min, and 95% ethanol 5 min, 85% ethanol for 5 min, 75% ethanol for 5 min, water for 5 min, and PBS washing 3 times for 5 min each time. An amount of 0.01 M PBS containing 0.5% Triton X-100 (pH = 7.2) was incubated for 20 min at RT and then washed 3 times in PBS for 5 min each time. A sodium citrate repair solution was used to repair antigens in a water bath at 85 °C for 45 min. Three percent H2O2 was used to eliminate endogenous peroxidase activity at RT for 20 min. An amount of 0.01 M PBS containing 5% goat serum (pH = 7.2) was incubated for 60 min at RT. Then 200 μL of 2 mAbs 79A11 and 69E2 at 8.95 μg/mL and 50 μg/mL, respectively, was added. The cover glasses were incubated overnight at 4 °C. Then 200 μL reagent 1 (polymer adjuvant) in the Dolink-2 Plus ® Polymer HRP Detection System was added and incubated at RT for 20 min and washed 3 times for 5 min each time. Then 200 μL reagent 2 (horseradish-labeled anti-mouse IgG polymer) was added and a DAB color development kit was used for coloration before the samples were observed under a microscope.
Western Blot was performed by using the IgM subtype mAb 79A11 and the IgG2a subtype mAb 69E2 to react with proteins extracted from CaSki and HeLa, respectively. A histidine-tagged human c-Myc (HIS-c-Myc) was used as a control to detect whether the mAbs 79A11 and 69E2 reacted with the HIS-tag. Detailed information is shown as follows: CaSki and HeLa cells were cultured and collected, and the total protein in CaSki and HeLa cells was extracted by using the Radioimmunoprecipitation Assay (RIPA) Lysis Buffer (Beyotime). Then the samples were separated by 12% SDS-PAGE, and the blots were transferred from the gel to nitrocellulose (NC) membrane. The NC membrane was blocked with 5% skim milk, and the primary antibodies 79A11 or 69E2 at 10 ug/mL were used to react with the protein on the NC membrane. Then the secondary antibody goat anti-mouse IgG-HRP was added, and photographed after exposure.
2.5. Characterization of Mouse anti-HPV16 E7-HIS Fusion Oncoprotein mAbs
The IgM subtype mAb 79A11 and IgG2a subtype 69E2 hybridoma cells were sent to Genscript (Nanjing, China) to determine the base sequence and predicted amino acid sequence. Briefly, total RNA was isolated from the hybridoma cells following the technical manual of TRIzol® Reagent (Ambion, Cat. No. 15596-026). Total RNA was then reverse-transcribed into cDNA using either isotype-specific anti-sense primers or universal primers following the technical manual of the PrimeScriptTM 1st Strand cDNA Synthesis Kit (Takara, Cat. No. 6110A). Antibody fragments of VH, VL, CH, and CL were amplified according to the GenScript standard operating procedure (SOP) for rapid amplification of cDNA ends (RACE). Amplified antibody fragments were cloned into a standard cloning vector separately. Colony PCR was performed to screen for clones with inserts of correct sizes. No less than 5 colonies with inserts of correct sizes were sequenced for each fragment. The sequences of the different clones were aligned and the consensus sequences were provided.
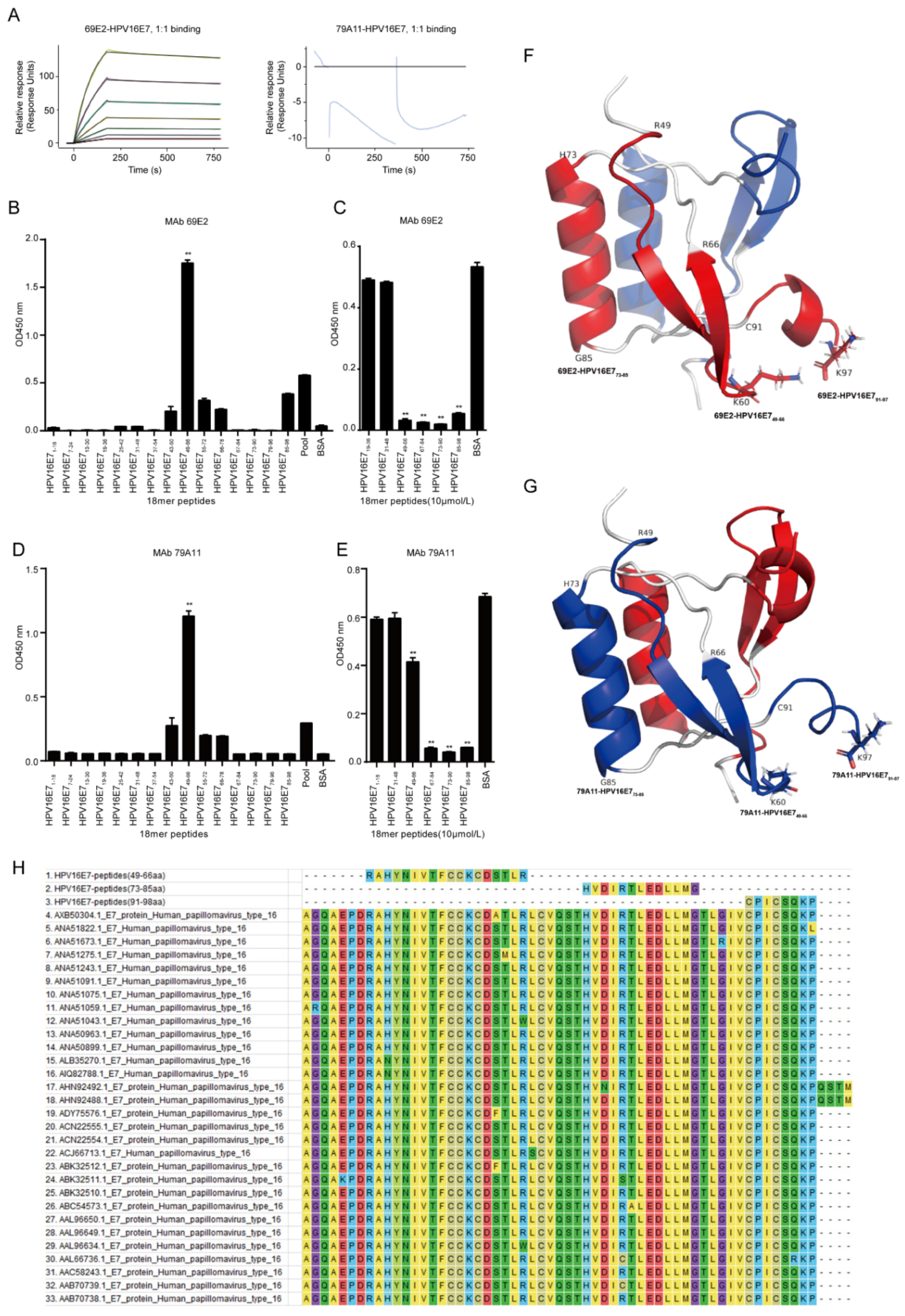
Affinity measurement was performed by GenScript Inc. (Nanjing, China). Briefly, the immobilization of HPV16 E7 was performed under 25 °C while 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffered saline with ethylene diamine tetraacetic acid (EDTA) and Polysorbate 20 (HBS-EP) buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA and 0.005% Tween-20) was used as the running buffer. The sensor chip surface of flow cells 1 and 2 were activated by freshly mixed 50 mM N-hydroxysuccinimide (NHS) and 200 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) for 420 s (10 µL/min). Afterwards, HPV16 E7 diluted in 10 mM NaAC (pH 4.5) was injected into flow cell 2 to achieve conjugation of the maximum (MAX) response unit respectively, while flow cell 1 was set as blank. After the amine coupling reaction, the remaining active coupling sites on the chip surface were blocked with a 420 s injection of 1 M ethanolamine hydrochloride. The assay was performed at 25 °C and the running buffer was HBS-EP: 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.05% Tween 20, pH 7.4 (GE Healthcare). Diluted 69E2 (IgG2a) and 79A11 (IgM) was injected over the surface as an association phase, followed by injecting running buffer as a dissociation phase. All the data were processed using the Biacore 8K evaluation software version 1.1 (GE Healthcare). Flow cell 1 and the blank injection of buffer in each cycle were used as double reference for response units (RUs) subtraction.
Dominant linear B-cell epitopes of mAbs was performed by indirect ELISA with 15 overlapping peptides that were 18 amino acids (aa) long with overlapping stretches of 6 aa. The HPV16 E7 protein sequence in the CaSki cell line on The National Center for Biotechnology Information (NCBI) website (ID: NC-001526.2) was used to design the overlapping peptides, which was subsequently synthesized by ChinaPeptides (Shanghai, China) with a purity greater than 95%. The synthesized overlapping peptides were dissolved in dimethyl sulfoxide (DMSO) to 1 mg/mL storage concentration and stored at −80 °C. The steps for indirect ELISA are shown as below: (1) Coating antigen: the bovine serum albumin (BSA) was set as a negative control protein, and a pool of 15 peptides mixed at a ratio of 1:1 was set as a positive control protein. The coating contents of 15 overlapping peptides, the negative control protein, and the positive control protein were all added as 5 μg per well. After incubating overnight at 4 °C, the plate was washed 4 times with PBST for 3 min each time; (2) Blocking: 250 μL 1.5% BSA in PBST was added and incubated at 37 °C for 2 h, and the plate was washed 3 times with PBST for 3 min each time; (3) Primary antibody incubation: the concentration of mAb 79A11 was 1.65 mg/mL, diluted 1:8000; the concentration of mAb 69E2 was 1.84 mg/mL, diluted 1:8000; 100 μL/well, 37 ° C for 1 h, washed with PBST for 4 times, 3 min for each time; (4) Secondary antibody incubation: anti-mouse IgG and HRP-linked antibody (Cat. No. 7076S, CST, Danvers, MA, USA) was diluted at 1:3000, 100 μL/well, 37 °C for 45 min, and then washed with PBST for 4 times, 3 min for each time; (5) TMB coloration: A and B solutions in the TMB two-component color development kit (Cat. No. PR1210, Solibao, Beijing, China) were diluted at 1:1, 100 μL/well, 37 °C for 5–8 min. Then 2 M H2SO4, 50 μL/well was added to stop the reaction and the optical density at 450 nm (OD450nm) value was detected.
Dominant linear B-cell epitopes of mAbs were screened by indirect competition ELISA, which consisted of 3 experiments: a criss-cross serial dilution analysis of indirect ELISA and 2 indirect competition ELISA experiments with overlapping peptides at 1 μM and 10 μM, respectively. Firstly, the coating concentration of antigen HPV16E7 fusion protien and response concentration of mAbs 79A11 and 69E2 were selected by using a criss-cross serial dilution analysis of indirect ELISA checkerboard experiment at an OD450nm value between 0.7 and 0.8. Then antigen HPV16E7 fusion protein were coated at optimized concentration, and double optimal concentrations of the mAbs were mixed with 15 overlapping peptides or BSA of 2 μM at a ratio of 1:1. After reacting at 25 °C for 30 min, the mixture was used to react with the antigen HPV16 E7 fusion protein, and overlapping peptides inhibited by the mAbs 79A11 and 69E2 were selected. The inhibition rate (IR) was used to determine the results of indirect competition ELISA. IR = ((OD450nm value of BSA − OD450nm value of overlapping peptide samples) × 100%)/(OD450nm value of BSA). IR ≥ 50% was considered positive. When the overlapping peptide was 1 μM, both mAbs 79A11 and 69E2 could inhibit part of the overlapping peptide, and the inhibition rates of most of the inhibited overlapping peptides were less than 50%, which is negative. Therefore, the concentration of overlapping peptides needed to be increased to 10 μM. The methods and steps of overlapping peptides at 10 μM were as follows: (1) Coating the antigen HPV16 E7 fusion protein: for mAb 79A11, when the coating concentration of the antigen HPV16 E7 protein was 1 μg/mL, the corresponding concentration of 79A11 mAb was 1 μg/mL; for the mAb 69E2, when the coating concentration of the antigen HPV16 E7 was 0.25 μg/mL, the corresponding concentration of the mAb 69E2 was 0.125 μg/mL. The plates were coated with 100 μL HPV16 E7 oncoprotein antigen at 4 °C overnight, and then washed with PBST 4 times, for 3 min each time; (2) Blocking: 1.0% BSA with PBST was prepared and incubated at 37 °C for 2 h, and then the plates were washed 3 times with PBST for 3 min each time; (3) Incubation with a mixture of primary antibody and overlapping peptides: mAbs 79A11 and 69E2 at 2 μg/mL and 0.25 μg/mL respectively were prepared, and several overlapping peptides inhibited by mAbs were selected, while 1–2 overlapping peptides that were not inhibited by the mAbs and BSA were used as negative controls. Afterwards, 2 μg/mL of mAb 79A11 and 0.25 μg/mL of mAb 69E2 were respectively mixed with 20 μM overlapping peptide, 20 mg/mL BSA, and PBST at 1:1, and incubated at 25 °C by shaking for 30 min, then these mixtures were added to the microplates coated with the HPV16 E7 fusion protein, 100 μL/well, and incubated at 37 °C for 1 h. Then the plates were washed 4 times with PBST for 3 min each time; (4) Secondary antibody incubation and TMB coloration were performed as indirect ELISA described above.
Using the HPV45 E7 dimer as a model in SWISS-MODEL homology modeling, a structural structure model of amino acids 46–97 of HPV16 E7 was obtained. Then the PyMOL (The PyMOL Molecular Graphics System 2.2.0, New York, NY, USA) was used to present the three-dimensional (3D) structural model of HPV16 E7. The software Molecular Evolutionary Genetics Analysis 7.0 (MEGA 7.0) was used to compare the amino acid sequences of the 3 exposed peptides of the HPV16 E7 protein with that of the other 30 HPV16 strains downloaded from the NCBI website.
2.6. Monoclonal Antibody Pairing
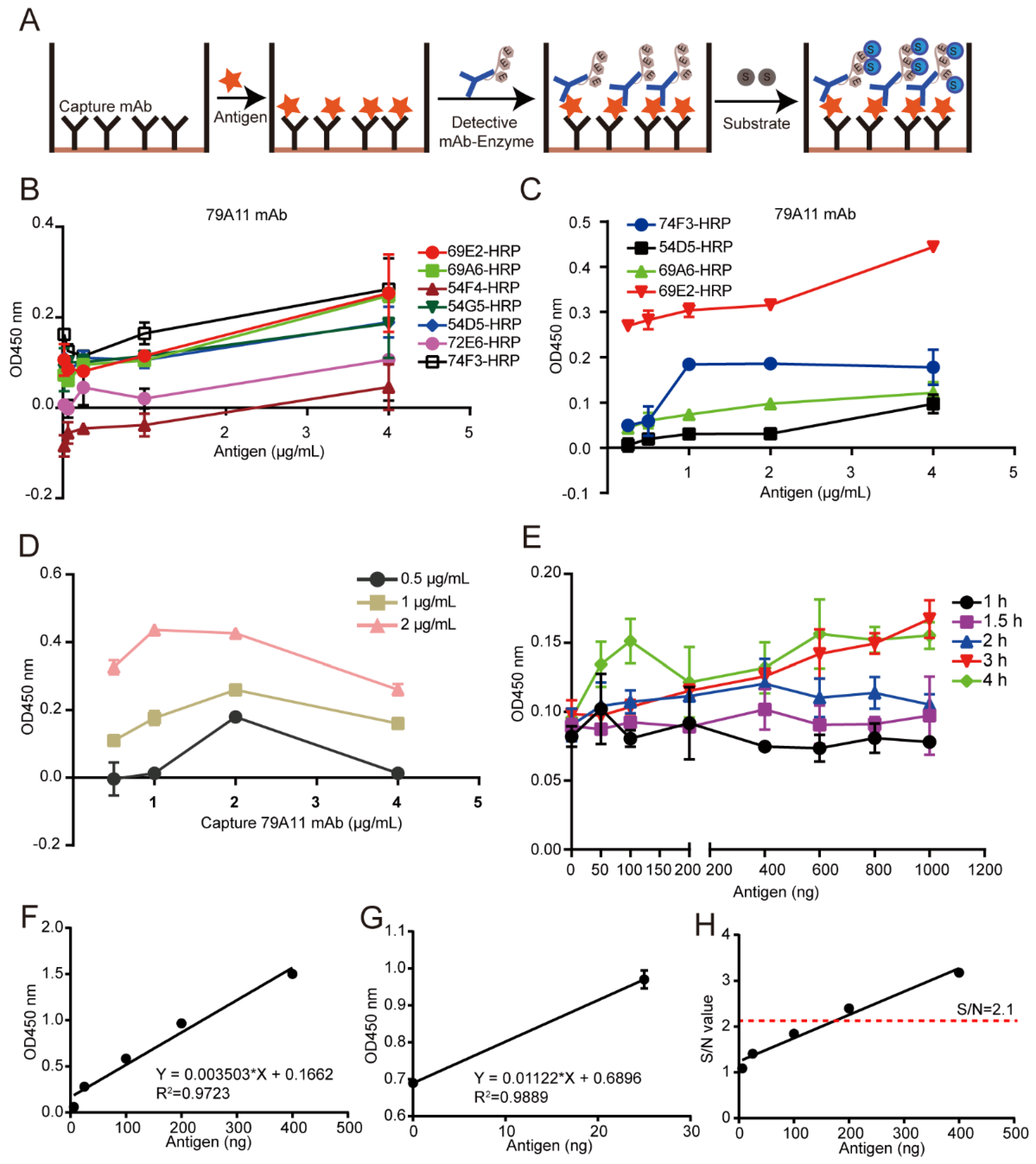
A double-antibody sandwich ELISA assay was used for pairing experiments. Unlabeled mAbs were used as capture antibodies, HRP-labeled mAbs as detection antibodies, and TMB substrate was used for coloration. The OD450 nm value was used to indirectly measure the antigen content. HRP was used to label 8 high effective mouse anti-HPV16 E7-HIS oncoprotein mAbs according to the instructions of the Peroxidase Labeling Kit-NH2 (Dojindo, Sahnghai, China). HRP-labeled mAbs were used as detection antibodies, and mAb pairing experiments were performed with unlabeled mAbs as capture antibodies. A total of 56 pairs were paired. The pair of mAbs with the strongest pairing signal was subjected to conventional double-antibody sandwich ELISA to quantitatively detect the lowest limitation of the HPV16 E7-HIS oncoprotein.
2.7. Establishment of a Double-Antibody Sandwich ELISA Method Based on HRP-Labeled mAb and TMB Detection System for Quantitative Detection of HPV16 E7-HIS Fusion Oncogenic Protein
Criss-cross serial dilution analysis was performed to obtain the optimal working concentration of the capture antibody, the optimized antigen concentration, and reaction time between the capture antibody and the antigen, thereby establishing a reference curve under the optimized conditions to obtain a linear regression equation. Briefly, a specified concentration of the 79A11 capture antibody was coated and incubated at 37 °C for 2 h, then transferred to 4 °C for 12–16 h, and then washed 3 times with PBST, each time for 3–5 min. Then 250 μL 5% skim milk dissolved in PBST was incubated at 37 °C for the indicated time for blocking, and the plate was washed 3 times with PBST for 3–5 min each time. One hundred milliliters of indicated concentrations of HPV16 E7 antigen were incubated for 2 h then washed 3 times with PBST for 3–5 min each time. One hundred milliliters of 1 μg/mL HRP-69E2 in PBST were incubated at 37 °C for 1 h, and washed 3 times with PBST, each time for 3–5 min. Then TMB was used for coloration and 2 M H2SO4 was used to stop the reaction. The OD450nm value was detected by a microplate reader.
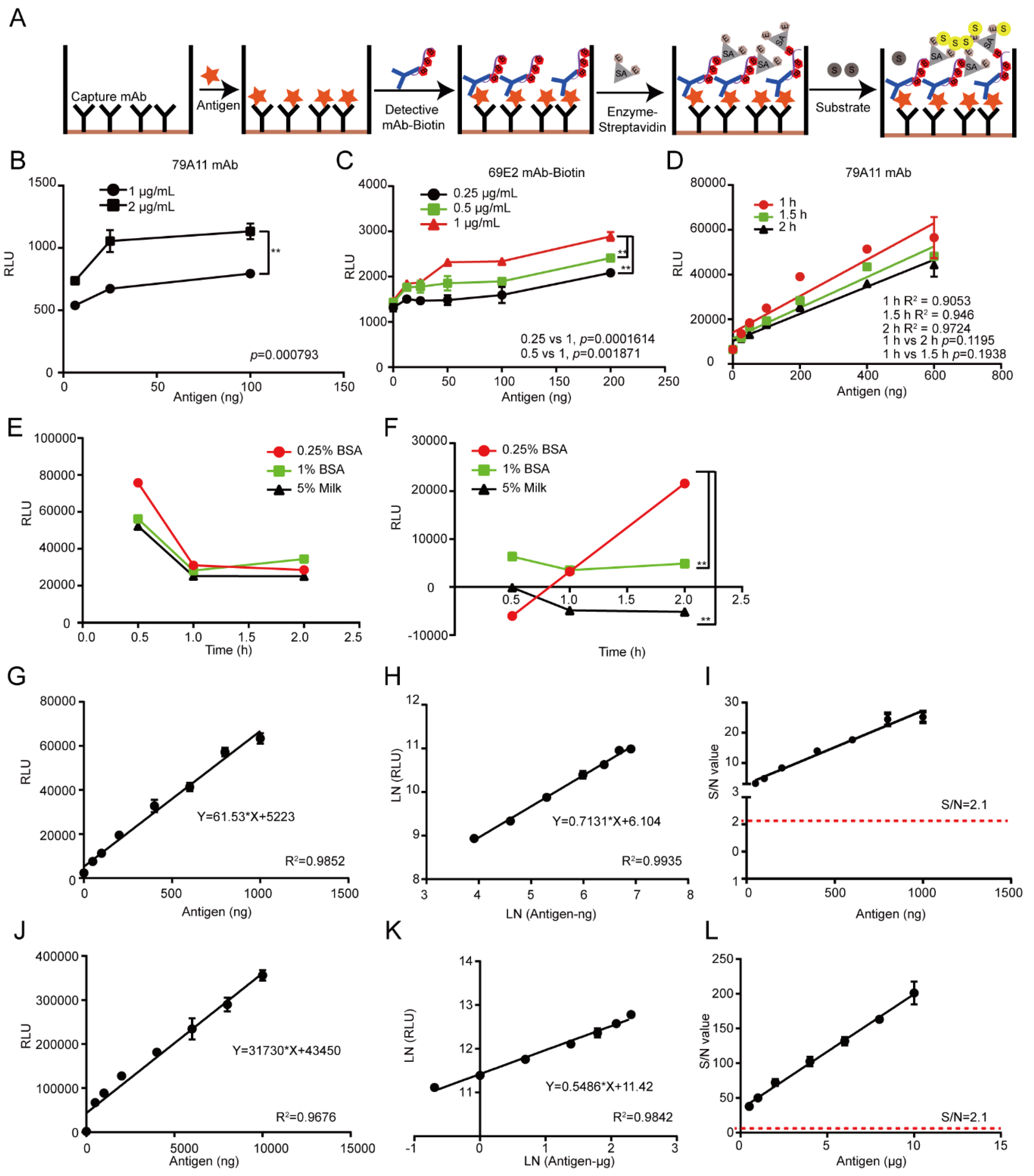
2.8. Establishment of the LSAB-ELISA and Luminol Detection System for Quantitative Detection of HPV16 E7-HIS Fusion Oncogenic Protein
According to the LK55 Biotin Labeling Kit-NH2 instructions, biotin was used to label the antibody 69E2 to obtain biotinylated detection antibody Biotin-69E2. Criss-cross serial dilution analysis by using a double-antibody sandwich ELISA was performed to optimize the optimal working concentrations of the capture mAb 79A11 and detection of the antibody Biotin-69E2, blocking conditions, and reaction time of the antigen HPV16 E7-HIS fusion oncoprotein with the antibody 79A11 (subtype IgM). The coating concentrations of the capture antibody 79A11 were 0.5, 1, 2, and 4 μg/mL, respectively. The concentrations of Biotin-69E2 were 0.25, 0.5, and 1 μg/mL, respectively. The blocking agents were 0.25% BSA, 1% BSA, and 5% skim milk, respectively. Blocking times were 30, 60, and 120 min, respectively. The reaction times of the antigen and capture antibody were 0.5, 1.5, and 2 h. Nine dilution points (0, 25, 50, 100, 200, 400, 600, 800, and 1000 ng per well) of the antigen were used to obtain the reference curve and determination coefficient. The upper detection limitation was determined by using 8 dilution points (0, 0.5, 1, 2, 4, 6, 8, and 10 μg of the antigen per well). The Chemiluminescence (relative light unit, RLU) of each reaction was detected by a microplate reader.
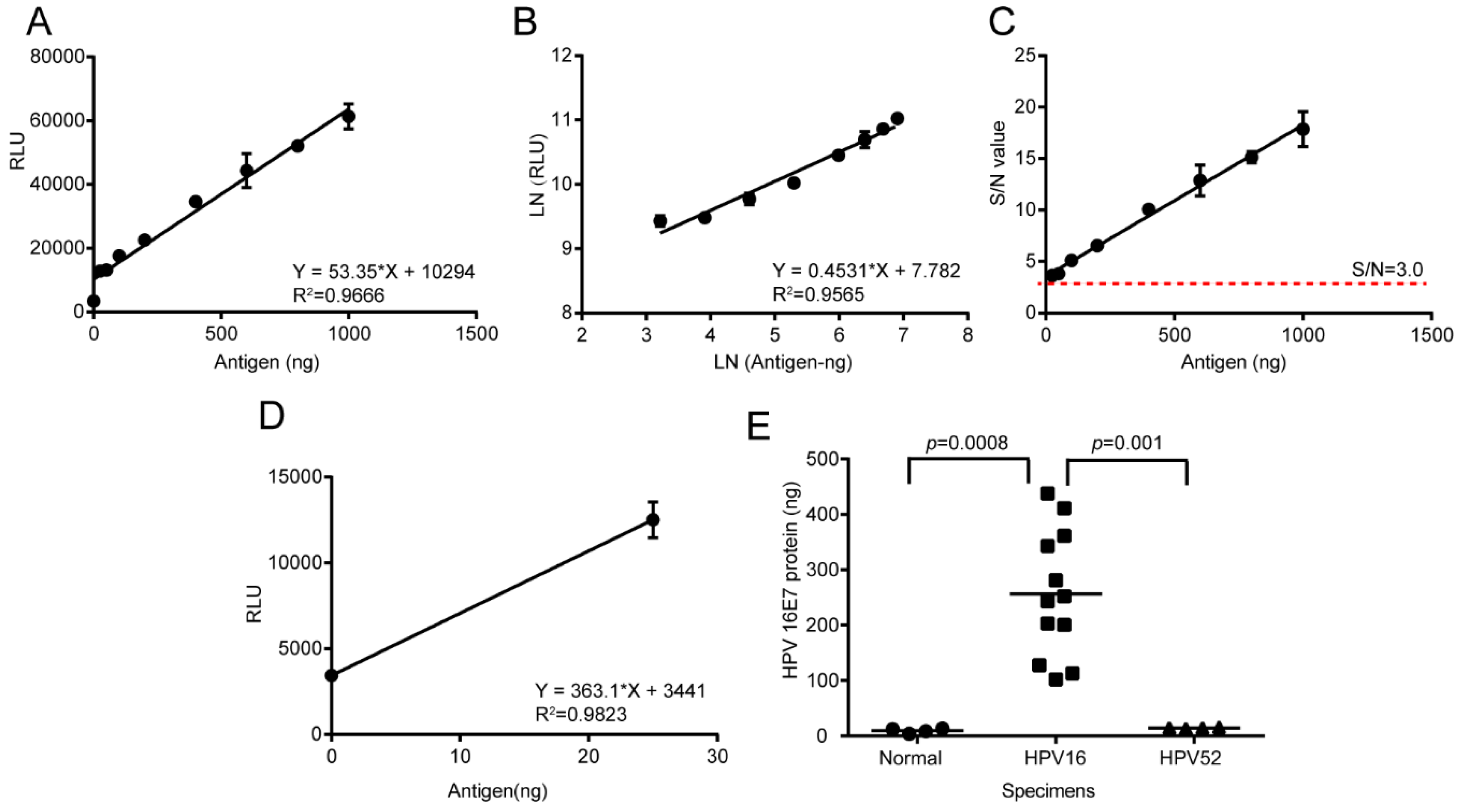
2.9. HPV16 E7 Oncoprotein Determination by Using the LSAB-ELISA and Luminol Detection System
The total protein of normal cervical tissue specimens, HPV16 positive cervical cancer specimens, and paracancerous specimens were extracted by using the RIPA Lysis Buffer. The Bicinchoninic Acid (BCA) Protein Assay Kit (Beyotime) was used to determine the total protein concentration. A reference curve was prepared under the optimal conditions and the HPV16 E7 oncogenic protein in 20 μg total protein of cervical cancer specimens was quantitatively detected by the established LSAB-ELISA and luminol detection system.
4. Discussion
The integration of HPV is an important factor during the malignant transformation of cervical epithelial cells [
17,
18]. HPV E6 and E7 are both oncogenic proteins [
19], but the HPV E7 oncogenic protein is more critical [
20]. Therefore, detection of HPV E7 is essential for the diagnosis of cervical cancer in the early stage [
21]. Recently, the HPV18 L1 ELISA detecting method and HPV16 E7-binding affibody molecules and nanobodies against the HPV16 E7 oncoprotein have been shown to have promising potential for the diagnosis of HPV-induced cancers [
22,
23,
24]. The goal of this research is to establish a method with high sensitivity for quantitative detection of the HPV16 E7 oncoprotein in cervical cancer tissues.
In this study, HPV16 E7-HIS fusion oncoprotein was successfully prepared, and the mAbs of mouse anti-HPV16 E7-HIS fusion oncoprotein were prepared and identified. The pair of mAbs with the strongest signals were used to establish a conventional double-antibody sandwich ELISA method and an LSAB-lumino-dual-antibody sandwich ELISA chemiluminescence immunoassay method. The results showed that the chemiluminescence immunoassay method was more sensitive than the conventional dual-antibody sandwich ELISA method in the quantitative detection of HPV16 E7-HIS fusion oncogenic protein. The clinical validity of this chemiluminescence immunoassay method was preliminarily verified with HPV16-positive cervical cancer specimens.
The reasons of why the established chemiluminescence immunoassay method based on LSAB-ELISA and luminol is better than the double-antibody sandwich ELISA based on HRP-labeled mAb are as follows: in the conventional double-antibody sandwich ELISA method, HRP directly labels the mAb without amplification, and the enzyme HRP itself is not sensitive, unstable, which causes the conventional double-antibody sandwich ELISA to have high background OD value and unsatisfactory sensitivity [
25]. The LSAB-ELISA is based on the principle of conventional ELISA and uses the height between biotin and streptavidin to amplify the effect and then replace the chromogenic substrate TMB with the chemiluminescence substrate luminol [
26,
27]. Additionally, dozens of biotin molecules are coupled to one antibody molecule without affecting the biological activity of antibody 69E2. The antigen, HPV16 E7-HIS fusion oncoprotein, binds to biotinylated mAb 69E2. Streptavidin in HRP-Streptavidin binds to four biotins in biotinylated antibodies Biotin-69E2. A streptavidin can bind multiple HRP enzymes, which catalyze chemiluminescent substrate luminol, thereby making the sensitivity of the chemiluminescent substrate higher than that of the chromogenic substrate, and the signal is amplified in multiple layers [
28,
29]. This method may be used to trace the antigen, antibodies, and receptors quantitatively and qualitatively.
The detection limit for the HPV16 E7 protein detected by the chemiluminescence immunoassay based on the LSAB-lumino-dual-antibody sandwich ELISA established in this study was 18.95 ng, which was 6.48 fold different from the detection limit of 122.92 ng of the dual-antibody sandwich ELISA method, indicating that the sensitivity of the luminescent immunoassay was higher than that of ordinary double-antibody sandwich ELISA. The S/N ratio of the HPV16 E7 protein content of the HPV16-positive cervical cancer tissues detected by the chemiluminescence immunoassay based on LSAB-ELISA-lumino-ELISA established in this subject was greater than 10, indicating that the signal was reliable and can also be detected. The S/N ratio of HPV16 E7 protein in the adjacent tissues was greater than 6.0, indicating that the method was reliable and effective, and provides some data references for further quantitative research.
To detect HPV16 E7 oncoproteins, corresponding anti-HPV16 E7 oncoprotein mAbs are required. To prepare the mAbs, the antigen HPV16 E7 oncoprotein must be successfully prepared. HPV cannot be cultivated outside of a host cell in a laboratory [
30]. Therefore, HPV16 E7 can only be prepared by genetic engineering techniques. Munger K. et al. showed that the HPV E7 protein often performs its function in the form of dimers or multimers in the zinc binding region [
31]. The molecular weight of the HPV16 E7 oncoprotein monomer is 12 kDa [
32], and the molecular weight of the HPV16 E7-HIS fusion oncoprotein prepared in this study was about 15–18 kDa, which may be caused by the protein’s negative charge [
14]. Because the HPV16 E7-HIS fusion oncoproteins cannot be completely equivalent to the natural HPV16 E7 oncoproteins in cervical cancer cell lines and cervical cancer tissues, the effectiveness and specificity of the mAbs prepared using the HPV16 E7-HIS fusion oncoproteins as antigens needed to be identified. Therefore, we used ICC, IF, IHC, Western blot, and the LSAB-ELISA reactions of cervical cancer tissues to verify the effectiveness and specificity of the mAbs against the HPV16 E7 protein.
The successful detection of HPV16 E7 oncoprotein greatly depends on successful mAb pairing. The most successful aspect of this study has been obtaining a pair of mAbs that can be paired and have a strong signal. The capture antibody is an IgM subtype and the detection antibody is an IgG2a subtype. The combination of the mAb with the capture antibody as an IgM subtype and the detection antibody as an IgG2a subtype is the biggest innovation of this study. The reasons for the successful analysis of the pairing are as follows: (1) The IgM mAb 79A11 of the paired mAbs was derived from the spleen cells of an immunized mouse, and the IgG2a mAb of the paired mAb was also derived from the same immunized mouse lymph node cells, which confirmed that the two mAbs are directed against two different epitopes in the same antigen HPV16 E7-HIS fusion oncoprotein. (2) Although the affinity of IgG and an antigen is stronger than IgM, as a pentamer, an IgM can bind to 10 antigens and can access cryptic antigens denied to IgG [
33]. IgM was used as a capture antibody in the double-antibody sandwich ELISA with less steric hindrance, which was more suitable for capturing antigens [
34]. (3) Further analysis determined that the mAb 79A11 subtype was indeed IgM, 69E2 was IgG2a, and the amino acid sequences of the base sequences of the light and heavy chains of the two mAbs were significantly different.
The specific epitopes of the mAbs 79A11 and 69E2 are very similar since they both contain three exposed peptides—HPV16 E749–66, HPV16 E773–85, and HPV16 E791–97. According to the data showing that the mAb 79A11 could be paired with the mAb 69E2 with strong pairing signals, it was proven that the specific epitopes of the mAbs 79A11 and 69E2 were in two different monomers of the same HPV16 E7 protein dimer. Combined with the result of the mAb 79A11 affinity test showing that the binding of mAb 79A11 to the antigen HPV16 E7 protein was eluted by high salt, it was speculated that the specific epitope peptides of mAb 79A11 were likely to contain lysines. This speculation was also supported by two pieces of evidence: (1) In the 3D crystal structure modeling diagram of the HPV16 E7 protein dimer, the 60th amino acid and the 97th amino acid of the HPV16E7 protein were lysines, and the positions of K60 and K97 were exposed and adjacent in space. (2) Indirect ELISA and indirect competition ELISA also showed that the mAb 79A11 specifically bound to the overlapping peptides HPV16 E749–66 and HPV16 E785–98, which contained K60 and K97, respectively. In contrast, the epitope of mAb 69E2 was less likely to contain lysine, because the affinity of mAb 69E2 to HPV16E7 protein was very high (5.60 × 10−9).
The detection method in this study can also optimize certain conditions to increase its sensitivity, such as using fresh antigen antibodies or obtaining the optimal conditions to prepare the reference curve in the fastest time. A better detection system is also needed to increase its sensitivity and reduce the background OD value. For example, HRP-Streptavidin can be replaced with alkaline phosphatase (AP)-Streptavidin [
35]. Because the sensitivity of AP is higher than that of HRP, the background OD value is lower and the signal is more stable [
36]. It is also possible to bind the capture antibody to magnetic beads. One limitation of this project is that the number of specimens is too small. In future research, more specimens should be used to verify the effectiveness of the method.
In conclusion, we successfully obtained two novel anti-HPV16 E7 oncoprotein mAbs and established an efficient method for the detection of anti-HPV16 E7 oncoprotein. The two novel anti-HPV16 E7 mAbs acquired in this study have many potential applications in experimental research and clinical diagnosis and therapy. Firstly, they can be developed as diagnostic kits, rapid diagnostic test strips, antibody-based biosensors or immunosensors, or other products that can detect the content of HPV16 E7 oncoprotein in cervical exfoliated cells, some of which may differentiate high-risk from low-risk [
37,
38,
39]. Secondly, the anti-HPV16 E7 protein mAbs can be transformed into human/mouse chimeric antibodies, or humanized antibodies through Fc modifications, which can be used for blocking the carcinogenic effect induced by the E7 oncogenic protein [
40,
41,
42]. Thirdly, HPV16 E7 human/mouse chimeric antibodies, or humanized HPV16 E7 antibodies can be conjugated with anti-cervical cancer drugs or radiolabels to develop radionuclide imaging agents for both the diagnosis and biological missiles for therapy of HPV16-positive neoplasms, including but not limited to, cervical cancers [
43,
44]. Fourthly, since the mAb MW is so large that it is difficult to be transported into cellular plasma, resulting in the mAb failing to block the intracellular E7 oncoprotein, the mAbs can provide some clues for the design of some intracellular antibodies (also known as single-chain variable fragments (scFvs)), which have been shown to have the potential for the treatment of HPV16-positive tumors [
45,
46,
47,
48]. Finally, these mAbs may also be used in experimental research. For instance, they can be used in immunoaffinity chromatography for purification of the HPV16 E7 antigen. Therefore, this study lies a foundation for the further exploration of antibody-based diagnosis and therapy for cervical cancer.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}