TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Constructs and Reagents
2.3. Sample Preparation, Western Blotting, and Immunoprecipitation
2.4. Immunofluorescence Assay
2.5. Plasmid Transfection
2.6. RNA Interference
2.7. CRISPR/Cas9
2.8. In Vitro Ubiquitination
2.9. Statistical Analysis
3. Results
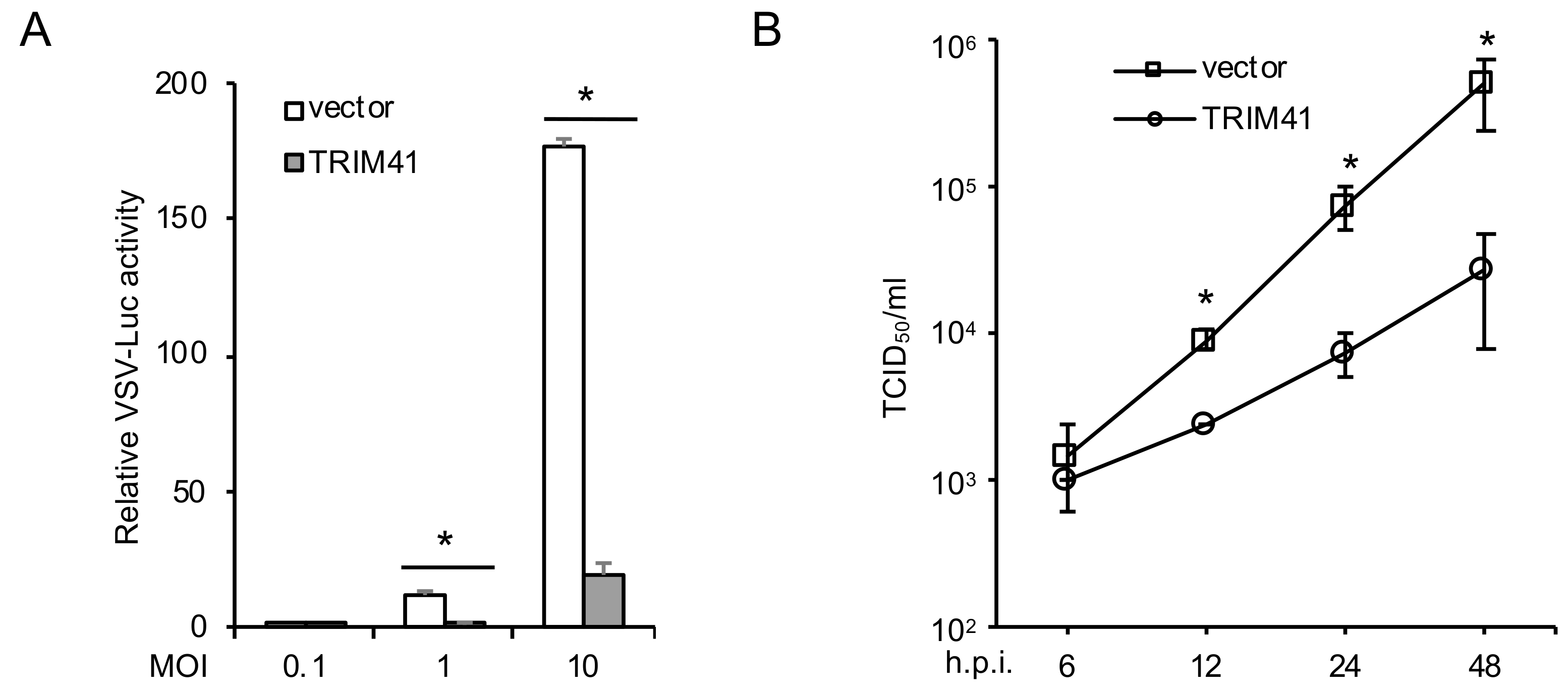
3.1. TRIM41 Restricts VSV Infection
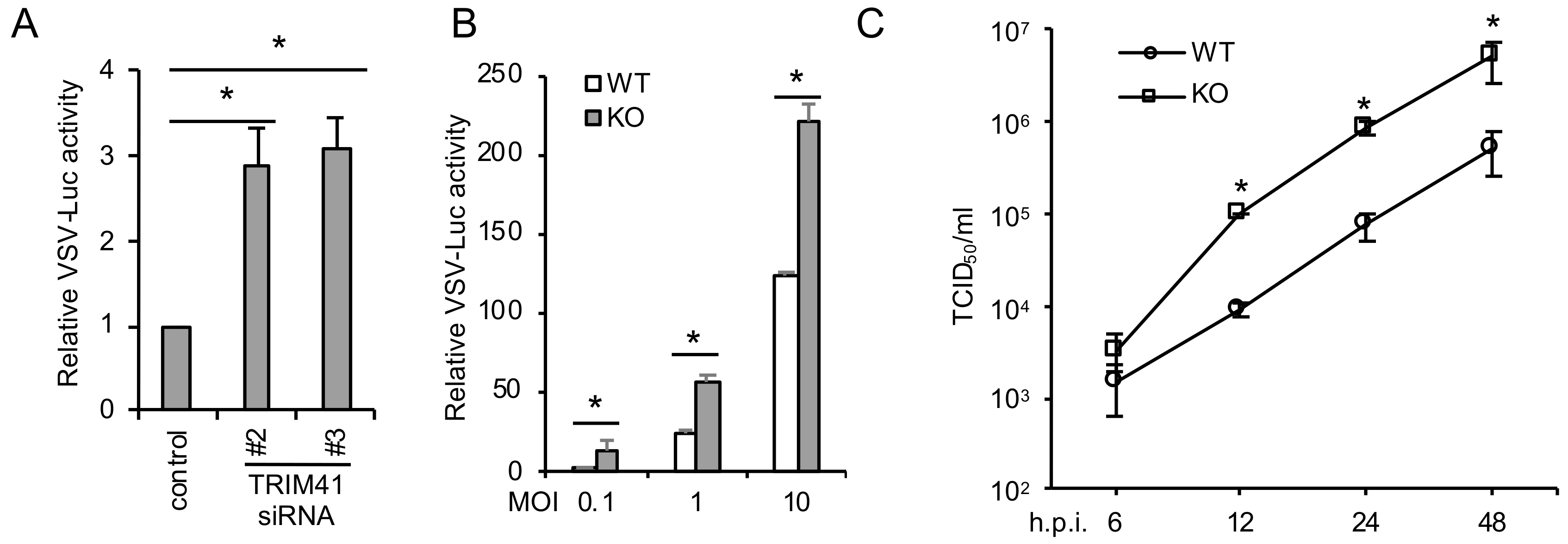
3.2. TRIM41 Deficiency Increases Host Susceptibility to VSV
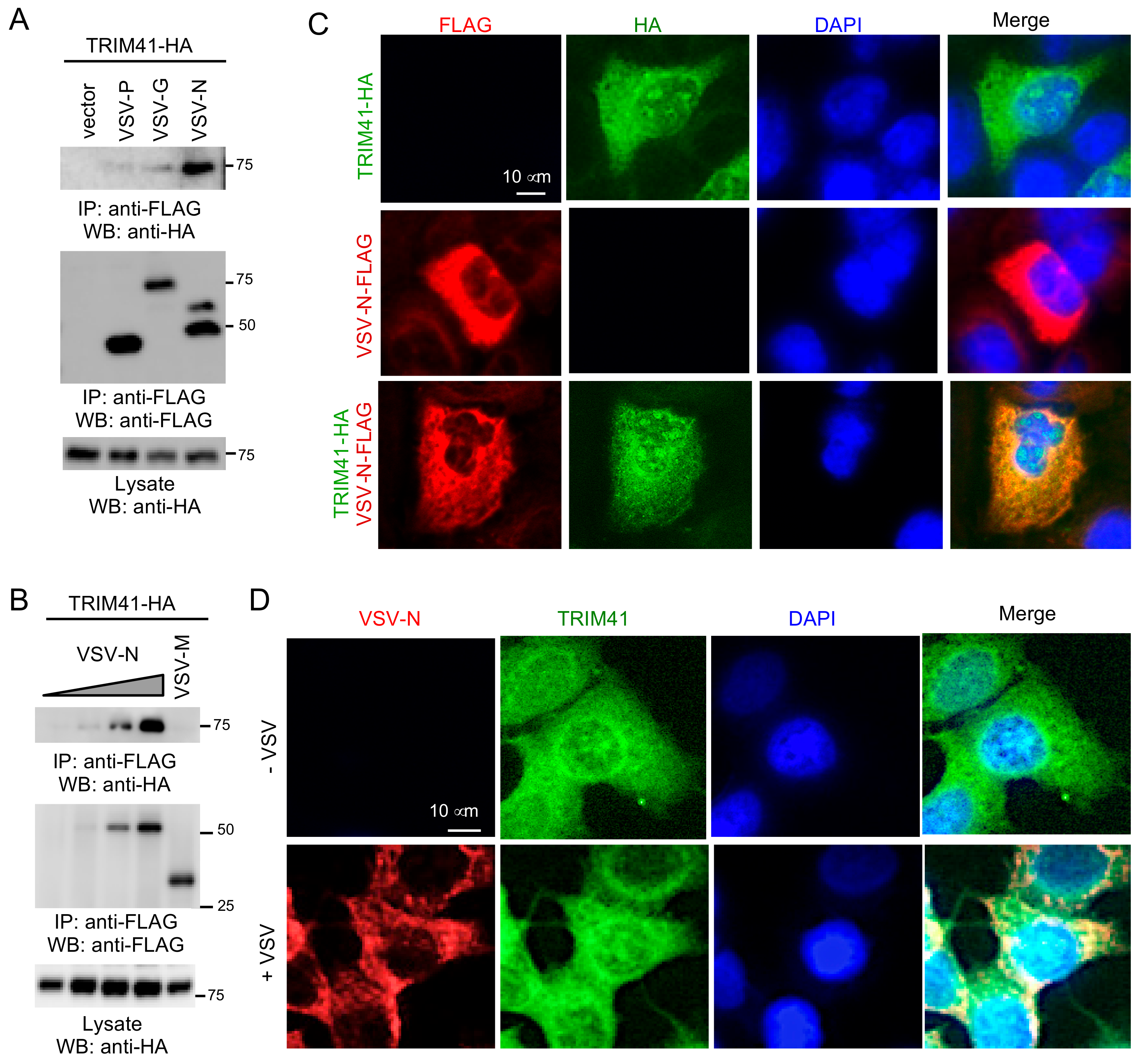
3.3. TRIM41 Interacts with the Nucleoprotein of VSV
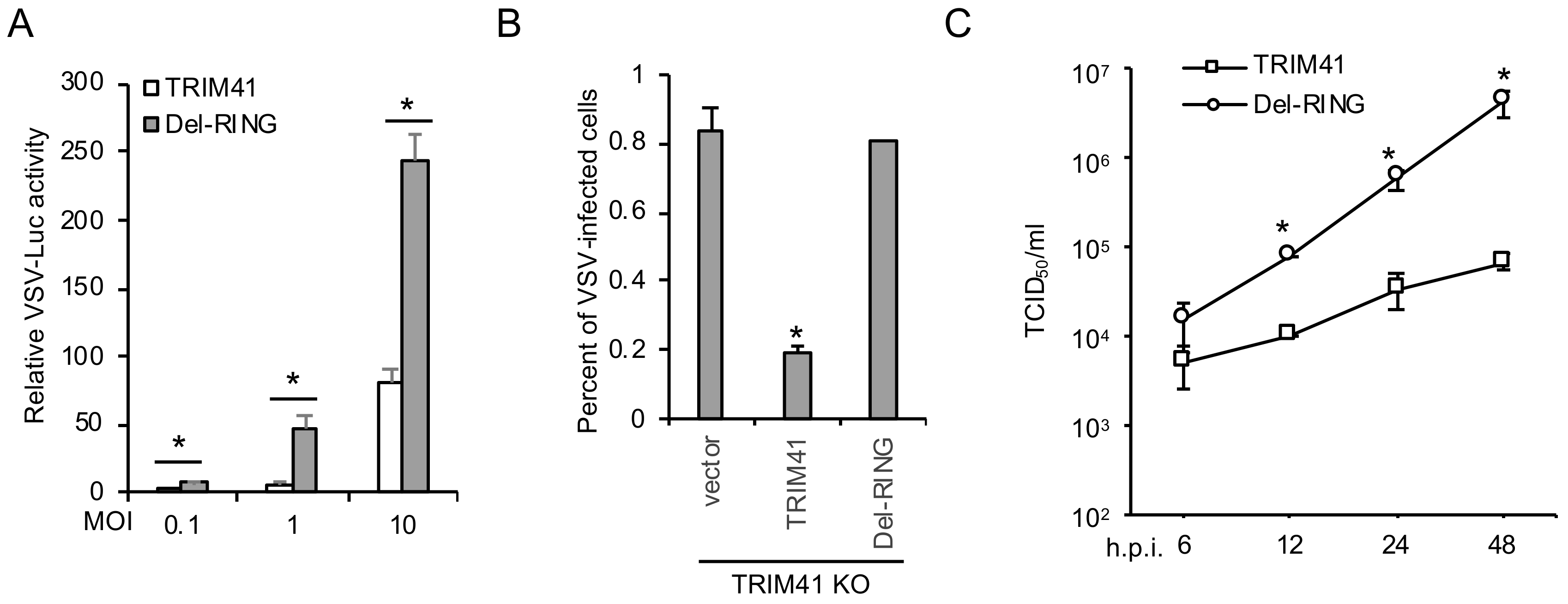
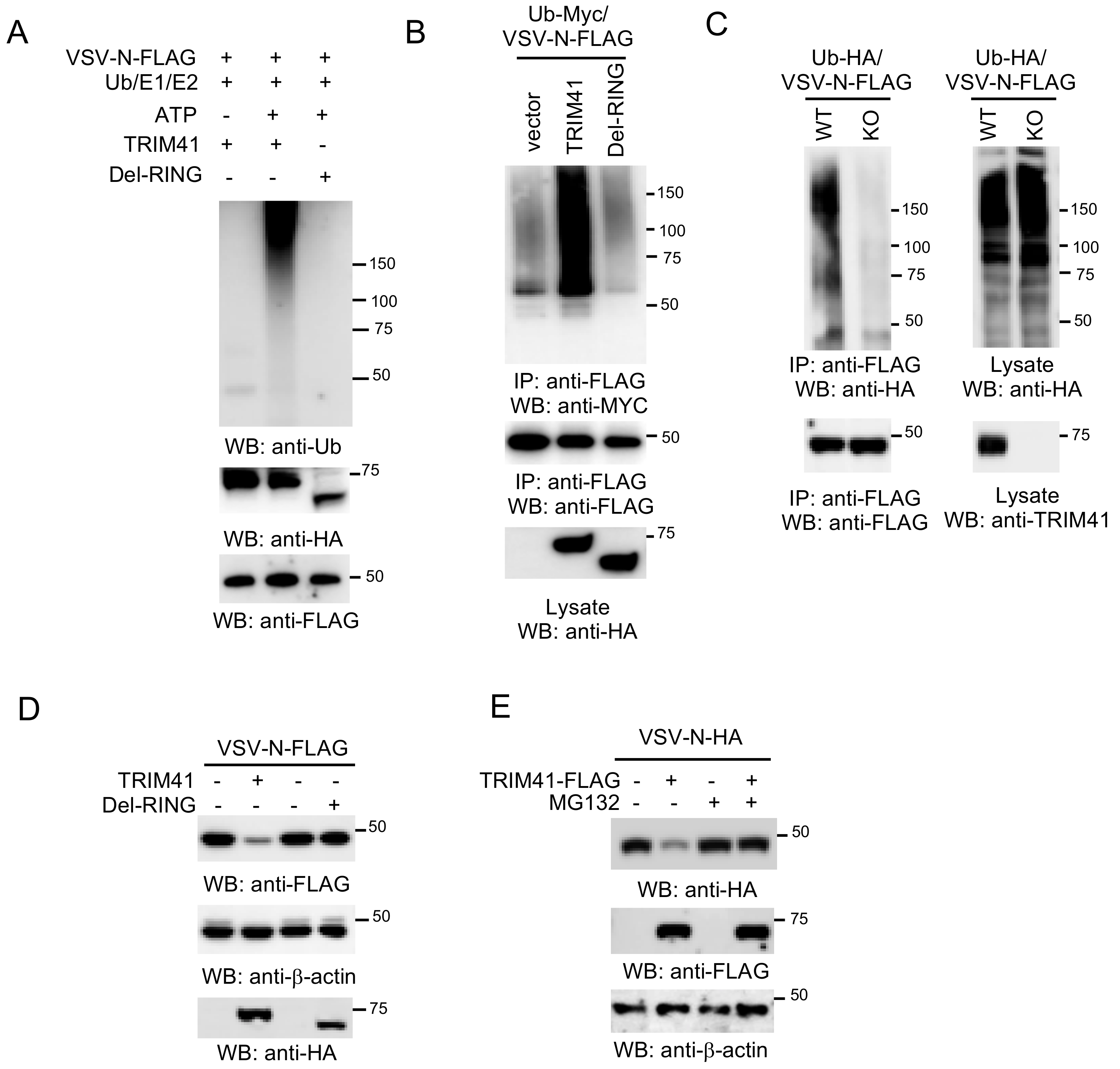
3.4. E3 Ligase Activity Is Required for TRIM41 Antiviral Function
3.5. TRIM41 Mediates the Ubiquitination and Degradation of VSV-N
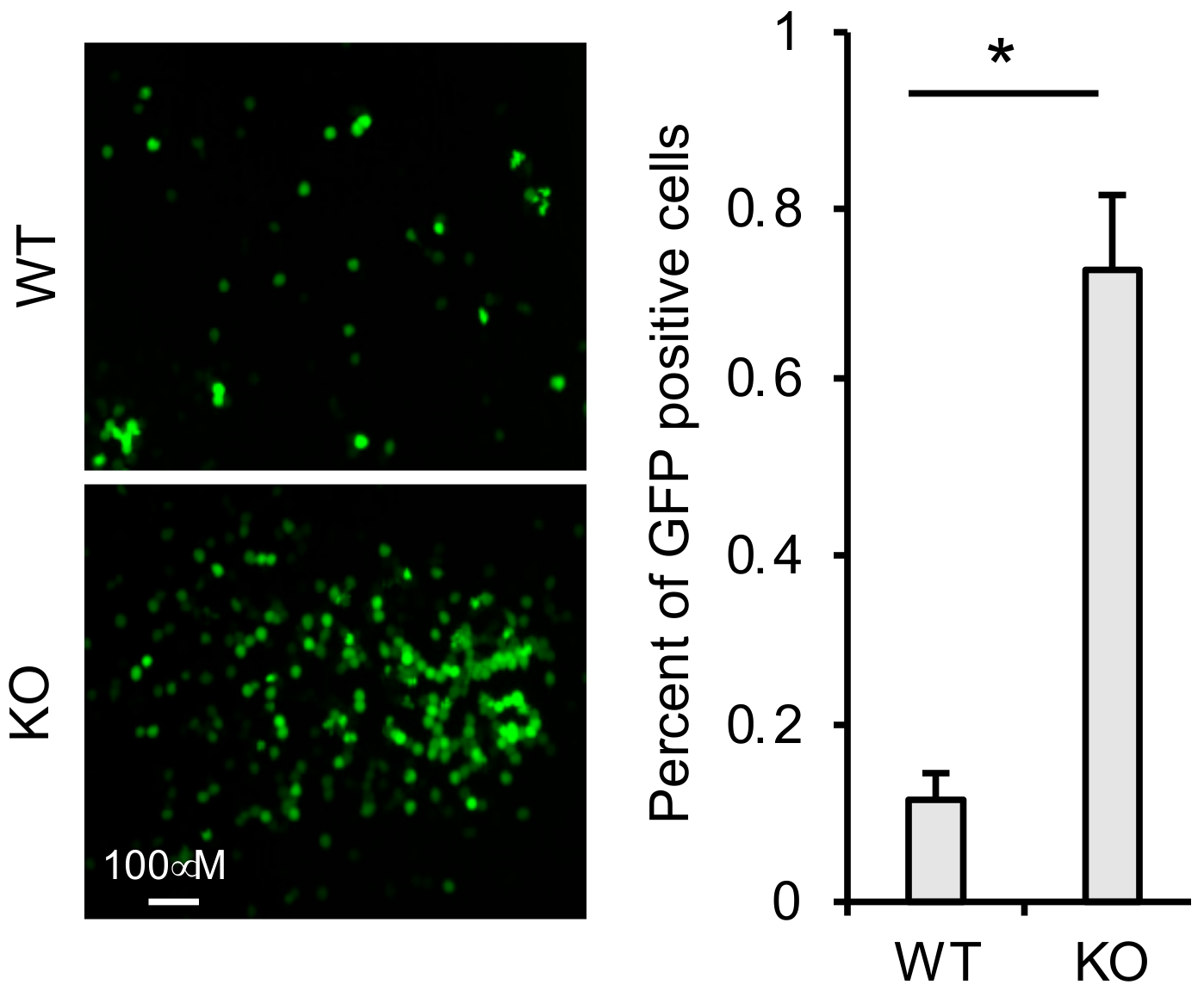
3.6. TRIM41 Restricts Incoming VSV Nucleocapsids
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Fu, Z.F. Genetic comparison of the rhabdoviruses from animals and plants. Curr. Top Microbiol. Immunol. 2005, 292, 1–24. [Google Scholar] [PubMed]
- Basak, S.; Mondal, A.; Polley, S.; Mukhopadhyay, S.; Chattopadhyay, D. Reviewing Chandipura: A vesiculovirus in human epidemics. Biosci. Rep. 2007, 27, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.; Conzelmann, K.K. Rhabdovirus evasion of the interferon system. J. Interferon Cytokine Res. 2009, 29, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Green, T.J.; Zhang, X.; Tsao, J.; Qiu, S. Conserved characteristics of the rhabdovirus nucleoprotein. Virus Res. 2007, 129, 246–251. [Google Scholar] [CrossRef]
- Luo, M.; Green, T.J.; Zhang, X.; Tsao, J.; Qiu, S. Structural comparisons of the nucleoprotein from three negative strand RNA virus families. Virol. J. 2007, 4, 72. [Google Scholar] [CrossRef][Green Version]
- Albertini, A.A.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W. Structural aspects of rabies virus replication. Cell Mol. Life Sci. 2008, 65, 282–294. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Chiang, J.J.; Sparrer, K.M.J.; Alam, S.L.; Chi, M.; Roganowicz, M.D.; Sankaran, B.; Gack, M.U.; Pornillos, O. Mechanism of TRIM25 catalytic activation in the antiviral RIG-I pathway. Cell Rep. 2016, 16, 1315–1325. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Patil, G.; Li, S. Tripartite motif proteins: An emerging antiviral protein family. Future Virol. 2019, 14, 107–122. [Google Scholar] [CrossRef]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 senses and restricts influenza a virus by ubiquitination of PB1 polymerase. PLoS Pathog 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, R.; Esmiol, S.; Lecine, P.; Mahfouz, B.; Hermant, A.; Nicoletti, C.; Parnis, S.; Perroy, J.; Borg, J.P.; Pascoe, L.; et al. Characterization and genetic analyses of new genes coding for NOD2 interacting proteins. PLoS ONE 2016, 11, e0165420. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.S.; Zhang, Z.Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X.M.; Li, T. Rinck-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Patil, G.; Zhao, M.; Song, K.; Hao, W.; Bouchereau, D.; Wang, L.; Li, S. TRIM41-mediated ubiquitination of nucleoprotein limits influenza a virus infection. J. Virol. 2018, 92, e00905-18. [Google Scholar] [CrossRef]
- Zhang, S.; Guo, J.T.; Wu, J.Z.; Yang, G. Identification and characterization of multiple trim proteins that inhibit hepatitis b virus transcription. PLoS ONE 2013, 8, e70001. [Google Scholar] [CrossRef]
- Wang, L.; Fu, B.; Li, W.; Patil, G.; Liu, L.; Dorf, M.E.; Li, S. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 2017, 8, 13876. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Sato, S.; Yamamoto, M.; Kaisho, T.; Sanjo, H.; Kawai, T.; Hoshino, K.; Takeda, K.; Akira, S. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 2004, 199, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- McWhirter, S.M.; Fitzgerald, K.A.; Rosains, J.; Rowe, D.C.; Golenbock, D.T.; Maniatis, T. IFN-regulatory factor 3-dependent gene expression is defective in TBK1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. USA 2004, 101, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Li, S. Influenza a virus-host protein interactions control viral pathogenesis. Int. J. Mol. Sci. 2017, 18, 1673. [Google Scholar] [CrossRef]
- Okada, A.; Iwatani, Y. APOBEC3G-mediated G-to-A hypermutation of the HIV-1 genome: The missing link in antiviral molecular mechanisms. Front. Microbiol. 2016, 7, 2027. [Google Scholar] [CrossRef]
- Yang, C.; Zhao, X.; Sun, D.; Yang, L.; Chong, C.; Pan, Y.; Chi, X.; Gao, Y.; Wang, M.; Shi, X.; et al. Interferon alpha (IFNα)-induced TRIM22 interrupts HCV replication by ubiquitinating NS5A. Cell Mol. Immunol. 2016, 13, 94–102. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Li, C.; Wu, Y.; Guo, L.; Peng, C.; Huang, Y.; Cheng, G.; Qin, F.X. TRIM14 inhibits hepatitis C virus infection by SPRY domain-dependent targeted degradation of the viral NS5A protein. Sci. Rep. 2016, 6, 32336. [Google Scholar] [CrossRef]
- Fan, W.; Wu, M.; Qian, S.; Zhou, Y.; Chen, H.; Li, X.; Qian, P. TRIM52 inhibits Japanese encephalitis virus replication by degrading the viral NS2A. Sci. Rep. 2016, 6, 33698. [Google Scholar] [CrossRef]
- Chelbi-Alix, M.K.; Quignon, F.; Pelicano, L.; Koken, M.H.; de The, H. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 1998, 72, 1043–1051. [Google Scholar] [CrossRef]
- Bonilla, W.V.; Pinschewer, D.D.; Klenerman, P.; Rousson, V.; Gaboli, M.; Pandolfi, P.P.; Zinkernagel, R.M.; Salvato, M.S.; Hengartner, H. Effects of promyelocytic leukemia protein on virus-host balance. J. Virol. 2002, 76, 3810–3818. [Google Scholar] [CrossRef] [PubMed]
- El Asmi, F.; Maroui, M.A.; Dutrieux, J.; Blondel, D.; Nisole, S.; Chelbi-Alix, M.K. Implication of PMLIV in both intrinsic and innate immunity. PLoS Pathog. 2014, 10, e1003975. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.C.; Chelbi-Alix, M.K. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 2011, 31, 145–158. [Google Scholar] [CrossRef]
- Wang, J.; Liu, B.; Wang, N.; Lee, Y.M.; Liu, C.; Li, K. TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 2011, 85, 3733–3745. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, N.L.; Wang, J.; Shi, P.Y.; Wang, T.; Miller, M.A.; Li, K. Overlapping and distinct molecular determinants dictating the antiviral activities of TRIM56 against flaviviruses and coronavirus. J. Virol. 2014, 88, 13821–13835. [Google Scholar] [CrossRef]
- Di Pietro, A.; Kajaste-Rudnitski, A.; Oteiza, A.; Nicora, L.; Towers, G.J.; Mechti, N.; Vicenzi, E. TRIM22 inhibits influenza a virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 2013, 87, 4523–4533. [Google Scholar] [CrossRef]
- Eldin, P.; Papon, L.; Oteiza, A.; Brocchi, E.; Lawson, T.G.; Mechti, N. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 2009, 90, 536–545. [Google Scholar] [CrossRef]
- Gao, B.; Duan, Z.; Xu, W.; Xiong, S. Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located ring domain. Hepatology 2009, 50, 424–433. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, G.; Xu, L.; Wu, Y.; Song, K.; Hao, W.; Hua, F.; Wang, L.; Li, S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection. Viruses 2020, 12, 131. https://doi.org/10.3390/v12020131
Patil G, Xu L, Wu Y, Song K, Hao W, Hua F, Wang L, Li S. TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection. Viruses. 2020; 12(2):131. https://doi.org/10.3390/v12020131
Chicago/Turabian StylePatil, Girish, Lingling Xu, Yakun Wu, Kun Song, Wenzhuo Hao, Fang Hua, Lingyan Wang, and Shitao Li. 2020. "TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection" Viruses 12, no. 2: 131. https://doi.org/10.3390/v12020131
APA StylePatil, G., Xu, L., Wu, Y., Song, K., Hao, W., Hua, F., Wang, L., & Li, S. (2020). TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection. Viruses, 12(2), 131. https://doi.org/10.3390/v12020131