
Enhanced Cell-Based Detection of Parvovirus B19V Infectious Units According to Cell Cycle Status

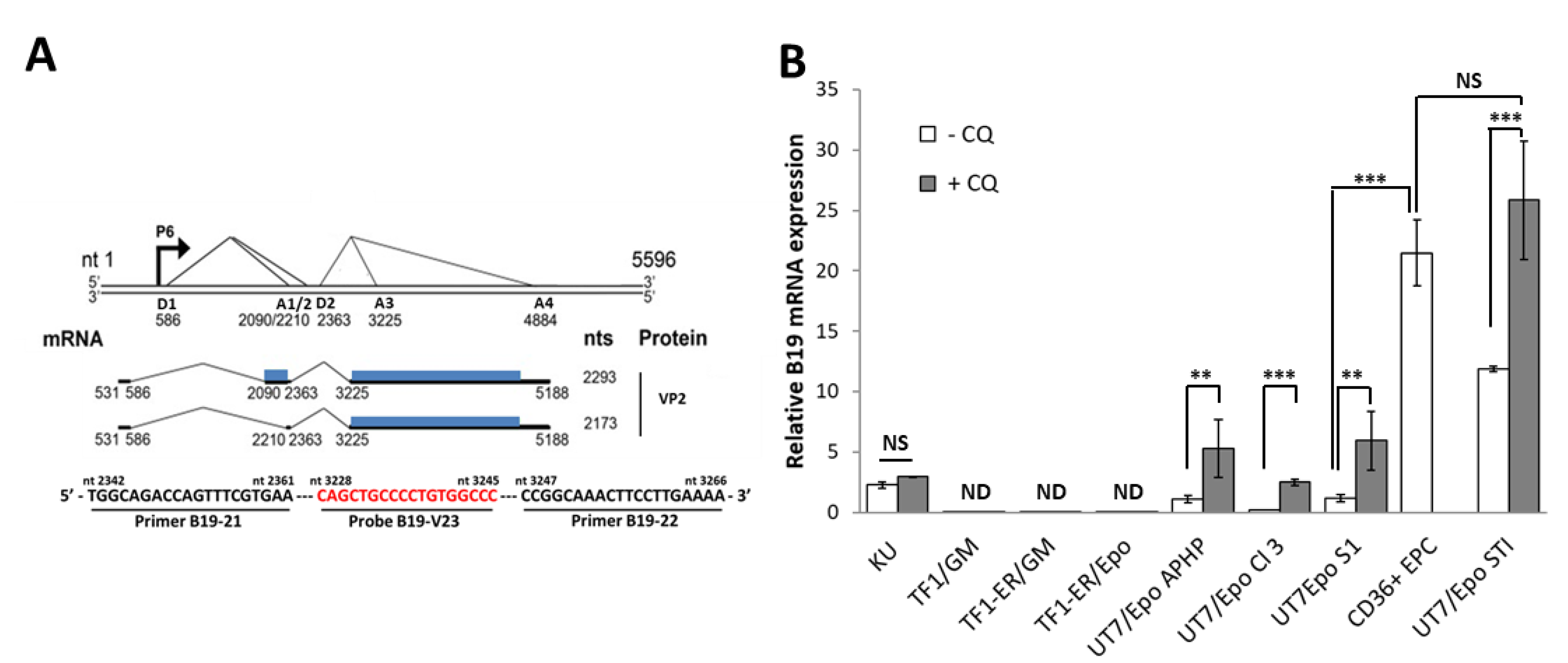{kind=link}
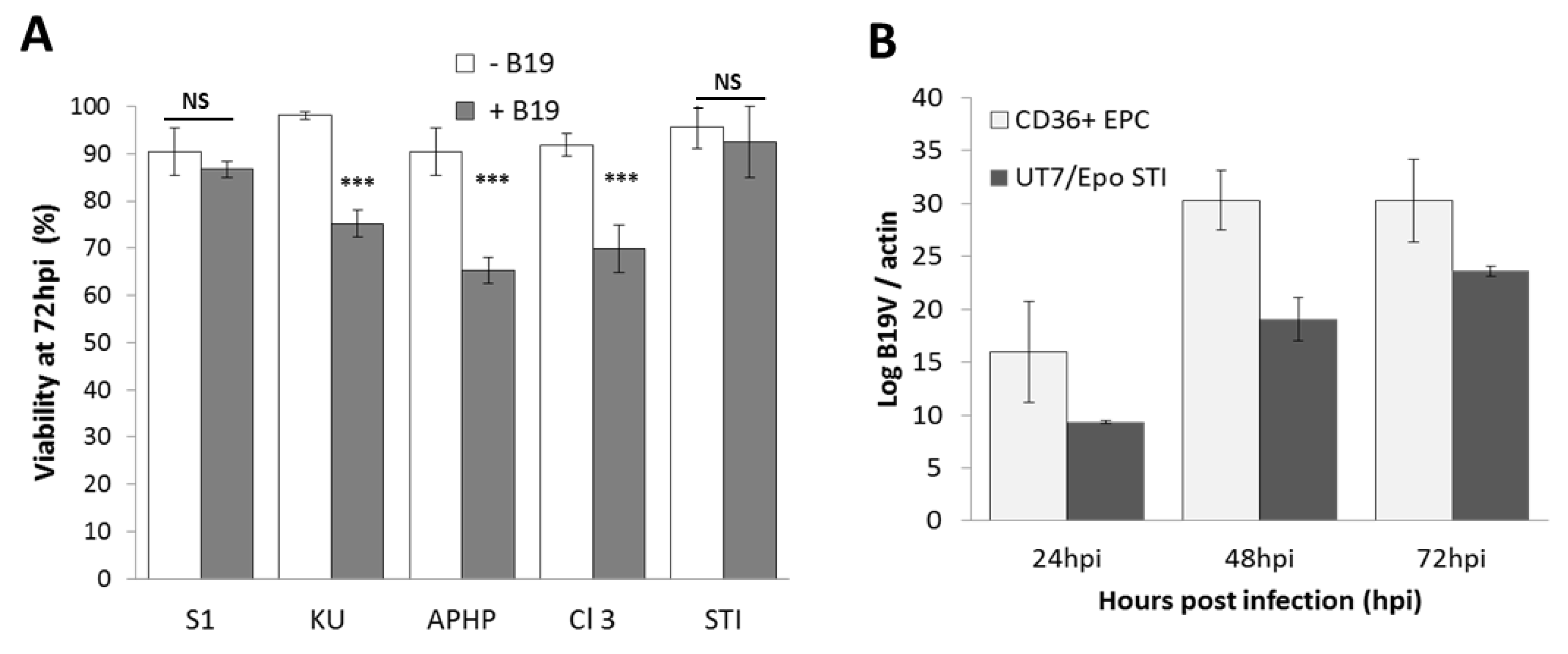{kind=link}
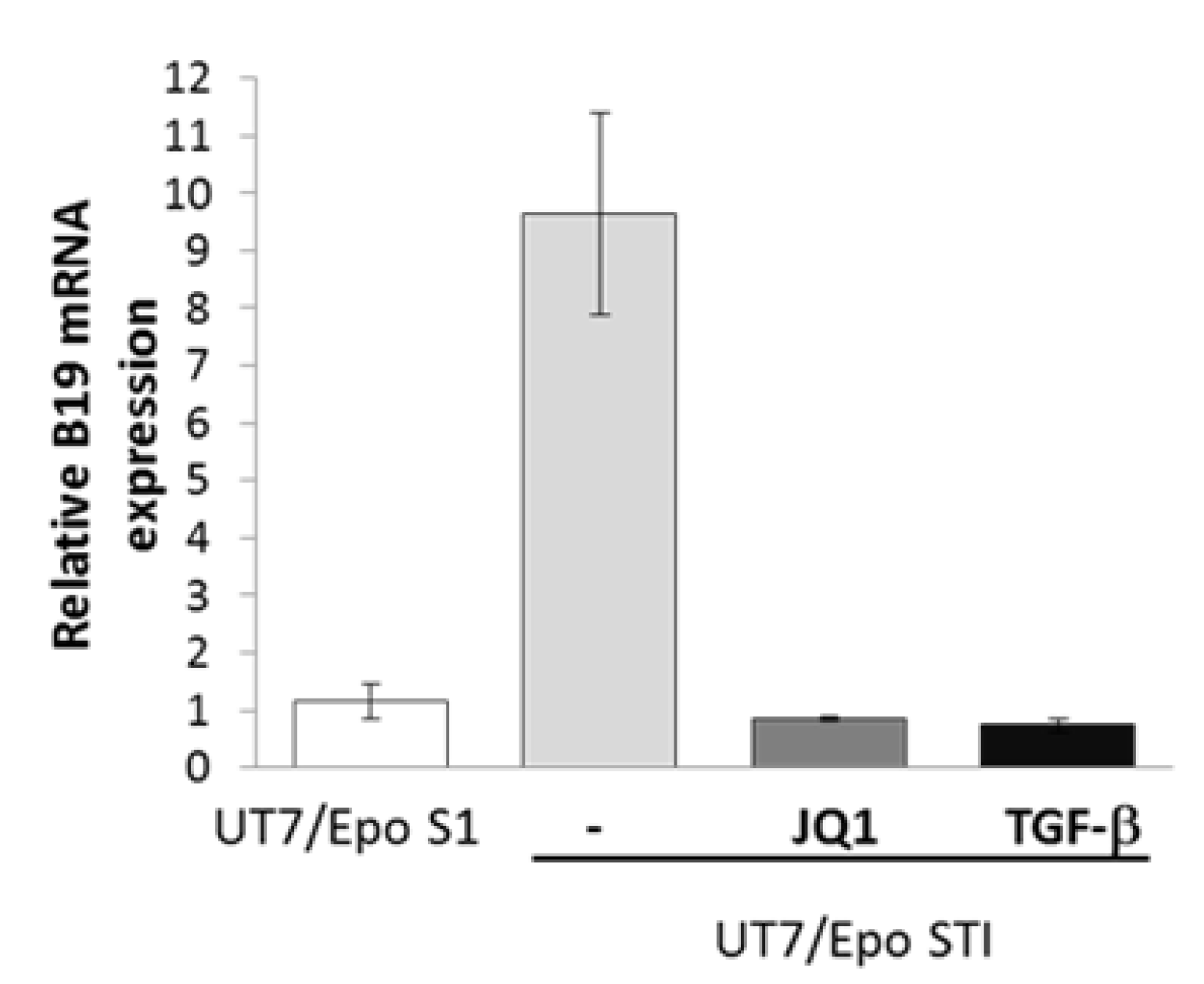{kind=link}
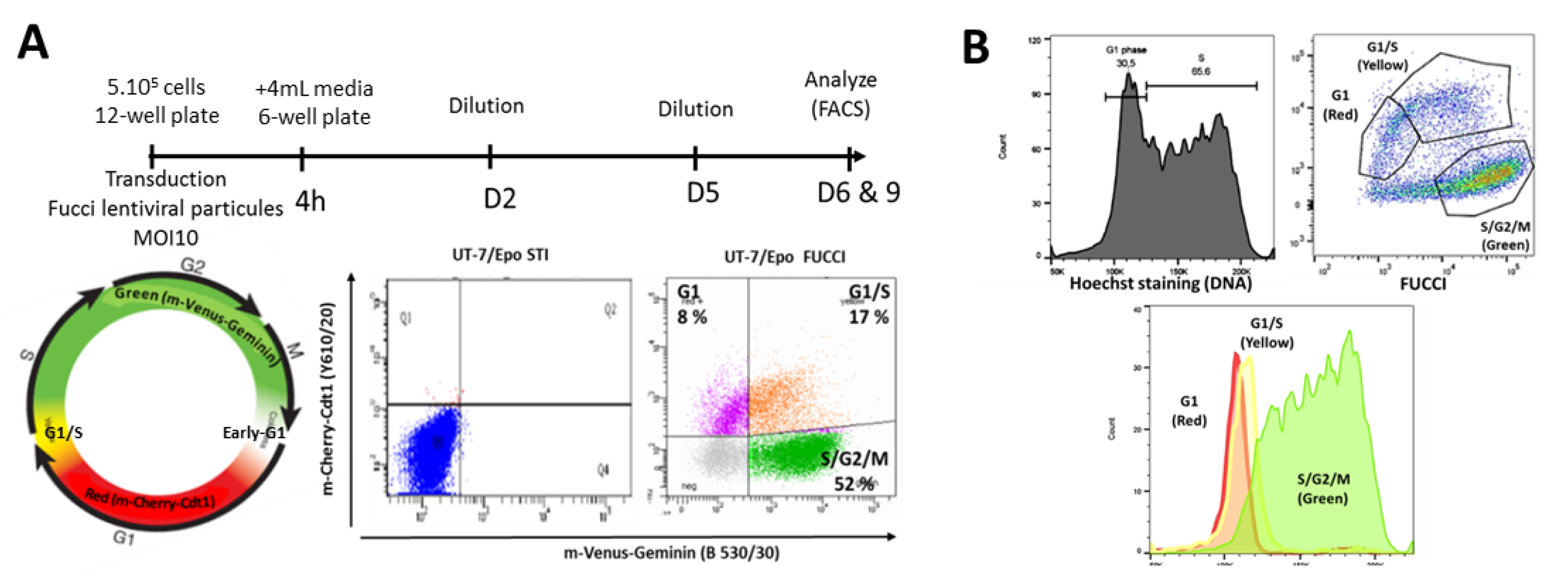{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. CD36+ Erythroid Progenitor Cell (EPC) Line Generation
2.3. B19 Virus Stock and Cell Inoculation
2.4. Fucci2a Lentivirus Production and Cell Transduction
2.5. UT7/Epo-FUCCI Clones Generation
2.6. Flow Cytometry Analysis of Cell Cycle Status
2.7. RNA Extraction and Duplex RT-qPCR
2.8. Statistical Analysis
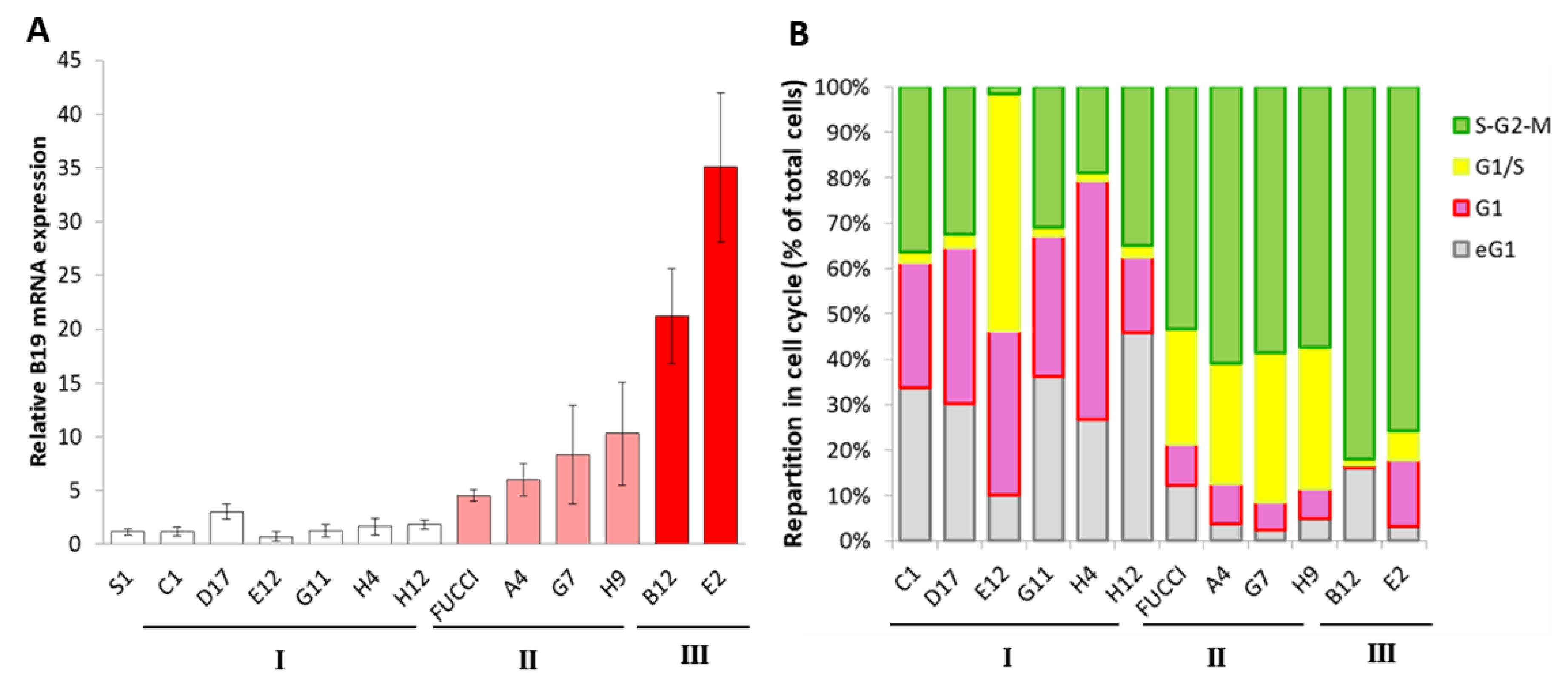
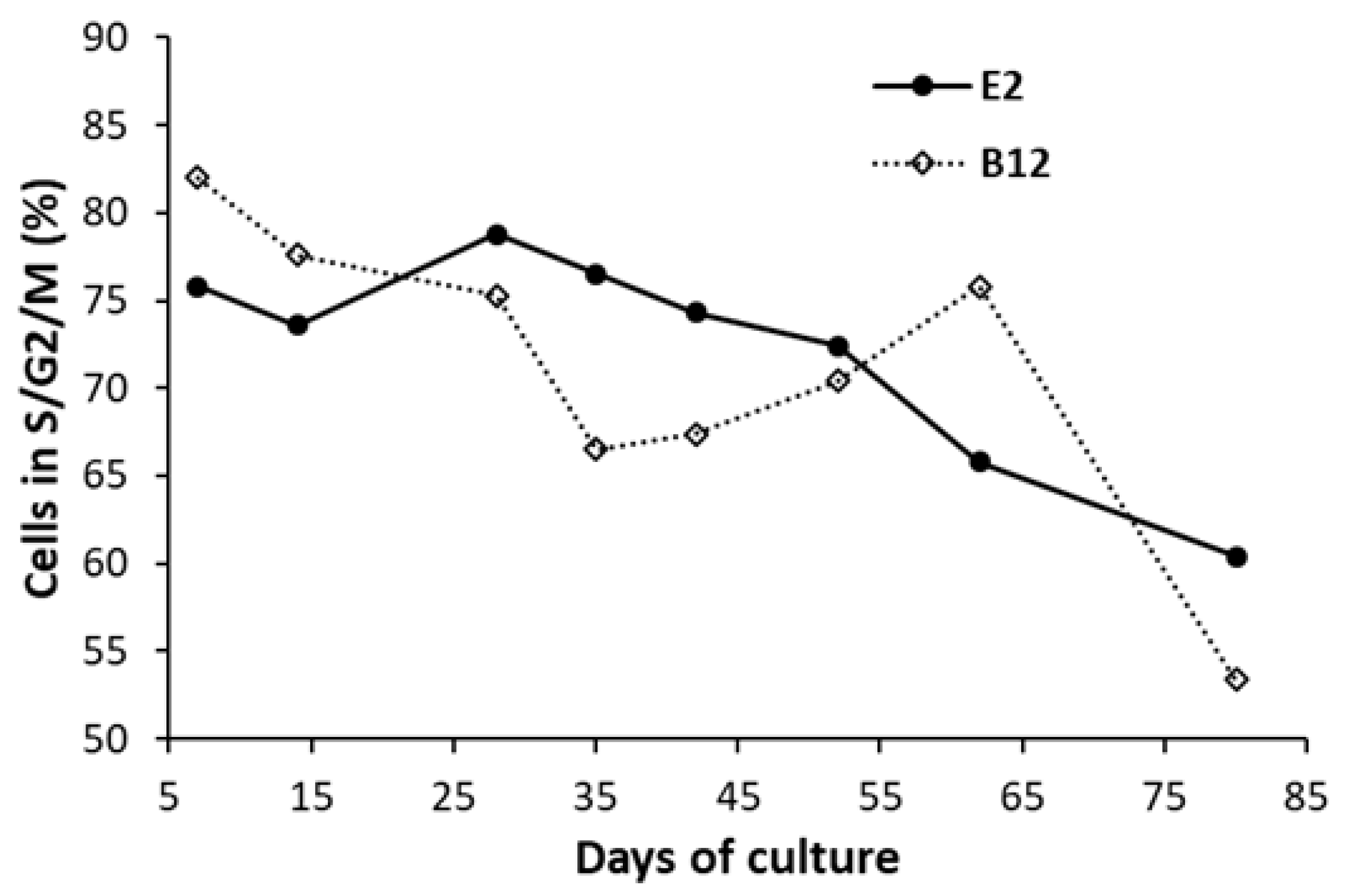
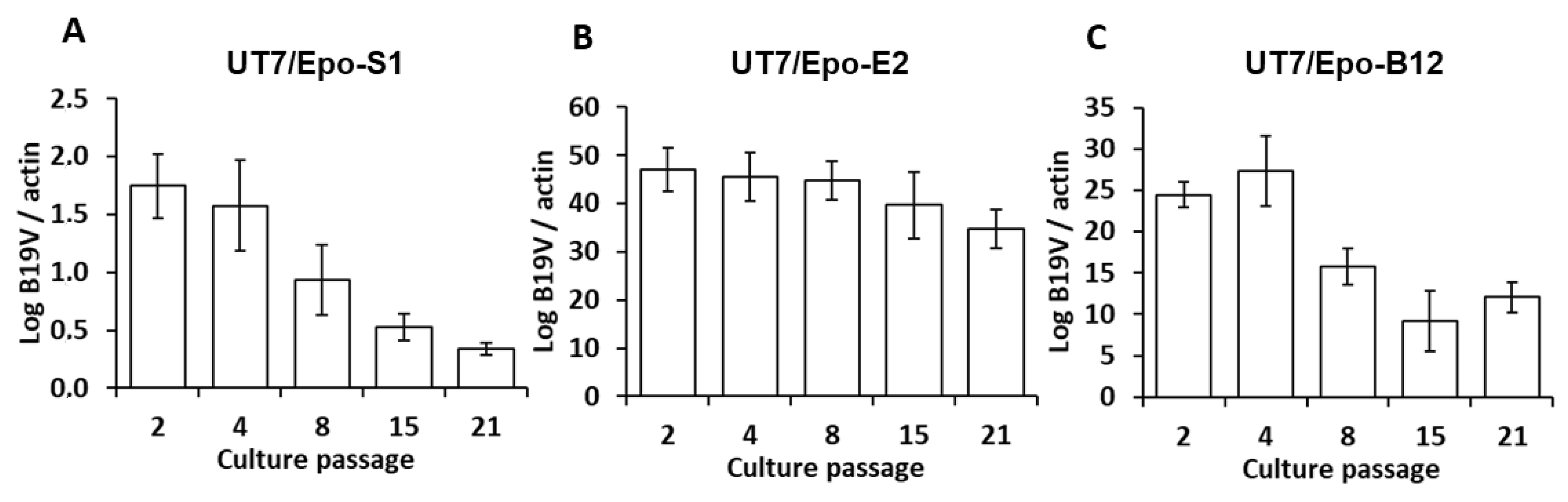
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Penzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Mietzsch, M.; Penzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362. [Google Scholar] [CrossRef]
- Brass, C.; Elliott, L.M.; Stevens, D.A. Academy rash. A probable epidemic of erythema infectiosum (‘fifth disease’). JAMA 1982, 248, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Jones, S.E.; Fisher-Hoch, S.P.; Lewis, E.; Hall, S.M.; Bartlett, C.L.; Cohen, B.J.; Mortimer, P.P.; Pereira, M.S. Human parvovirus, the cause of erythema infectiosum (fifth disease)? Lancet 1983, 1, 1378. [Google Scholar] [CrossRef]
- Hosszu, E.; Sallai, A. Human parvovirus B19 infection in a child suffering from chronic arthritis. Orv. Hetil. 1997, 138, 611–613. [Google Scholar] [PubMed]
- Frickhofen, N.; Chen, Z.J.; Young, N.S.; Cohen, B.J.; Heimpel, H.; Abkowitz, J.L. Parvovirus B19 as a cause of acquired chronic pure red cell aplasia. Br. J. Haematol. 1994, 87, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, S.S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell Infect. Microbiol. 2018, 8, 166. [Google Scholar] [CrossRef]
- Young, N.; Harrison, M.; Moore, J.; Mortimer, P.; Humphries, R.K. Direct demonstration of the human parvovirus in erythroid progenitor cells infected in vitro. J. Clin. Investig. 1984, 74, 2024–2032. [Google Scholar] [CrossRef]
- Kurtzman, G.J.; Ozawa, K.; Cohen, B.; Hanson, G.; Oseas, R.; Young, N.S. Chronic bone marrow failure due to persistent B19 parvovirus infection. N. Engl. J. Med. 1987, 317, 287–294. [Google Scholar] [CrossRef]
- Duncan, J.R.; Potter, C.B.; Cappellini, M.D.; Kurtz, J.B.; Anderson, M.J.; Weatherall, D.J. Aplastic crisis due to parvovirus infection in pyruvate kinase deficiency. Lancet 1983, 2, 14–16. [Google Scholar] [CrossRef]
- Bertrand, J.; Dennis, M.; Cutler, T. It’s Complicated: Parvovirus B19 in Thalassemia. Am. J. Med. 2017, 130, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Obeid Mohamed, S.O.; Osman Mohamed, E.M.; Ahmed Osman, A.A.; Abdellatif MohamedElmugadam, F.A.; Abdalla Ibrahim, G.A. A Meta-Analysis on the Seroprevalence of Parvovirus B19 among Patients with Sickle Cell Disease. Biomed. Res. Int. 2019, 2019, 2757450. [Google Scholar] [CrossRef] [PubMed]
- Obeid, K.M. Infections with DNA Viruses, Adenovirus, Polyomaviruses, and Parvovirus B19 in Hematopoietic Stem Cell Transplant Recipients and Patients with Hematologic Malignancies. Infect. Dis. Clin. N. Am. 2019, 33, 501–521. [Google Scholar] [CrossRef]
- Tolfvenstam, T.; Papadogiannakis, N.; Norbeck, O.; Petersson, K.; Broliden, K. Frequency of human parvovirus B19 infection in intrauterine fetal death. Lancet 2001, 357, 1494–1497. [Google Scholar] [CrossRef]
- Brown, T.; Anand, A.; Ritchie, L.D.; Clewley, J.P.; Reid, T.M. Intrauterine parvovirus infection associated with hydrops fetalis. Lancet 1984, 2, 1033–1034. [Google Scholar] [CrossRef]
- Satake, M.; Hoshi, Y.; Taira, R.; Momose, S.Y.; Hino, S.; Tadokoro, K. Symptomatic parvovirus B19 infection caused by blood component transfusion. Transfusion 2011, 51, 1887–1895. [Google Scholar] [CrossRef]
- Nagaharu, K.; Sugimoto, Y.; Hoshi, Y.; Yamaguchi, T.; Ito, R.; Matsubayashi, K.; Kawakami, K.; Ohishi, K. Persistent symptomatic parvovirus B19 infection with severe thrombocytopenia transmitted by red blood cell transfusion containing low parvovirus B19 DNA levels. Transfusion 2017, 57, 1414–1418. [Google Scholar] [CrossRef]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Calizzani, G.; Candura, F.; Liumbruno, G.M.; Grazzini, G. Human Parvovirus B19 and blood product safety: A tale of twenty years of improvements. Blood Transfus. 2015, 13, 184–196. [Google Scholar] [CrossRef]
- Baylis, S.A.; Wallace, P.; McCulloch, E.; Niesters, H.G.M.; Nubling, C.M. Standardization of Nucleic Acid Tests: The Approach of the World Health Organization. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef]
- Molenaar-de Backer, M.W.; Russcher, A.; Kroes, A.C.; Koppelman, M.H.; Lanfermeijer, M.; Zaaijer, H.L. Detection of parvovirus B19 DNA in blood: Viruses or DNA remnants? J. Clin. Virol. 2016, 84, 19–23. [Google Scholar] [CrossRef]
- Baylis, S.A.; Ma, L.; Padley, D.J.; Heath, A.B.; Yu, M.W.; Grp, C.S. Collaborative study to establish a World Health Organization International genotype panel for parvovirus B19 DNA nucleic acid amplification technology (NAT)-based assays. Vox Sang. 2012, 102, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Brown, K.E. Development of an improved method of detection of infectious parvovirus B19. J. Clin. Virol. 2006, 35, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.J.; Ardura, M.I.; The AST Infectious Diseases Community of Practice. Human parvovirus B19 in solid organ transplantation: Guidelines from the American society of transplantation infectious diseases community of practice. Clin. Transplant. 2019, 33, e13535. [Google Scholar] [CrossRef]
- Sundin, M.; Lindblom, A.; Orvell, C.; Barrett, A.J.; Sundberg, B.; Watz, E.; Wikman, A.; Broliden, K.; Le Blanc, K. Persistence of human parvovirus B19 in multipotent mesenchymal stromal cells expressing the erythrocyte P antigen: Implications for transplantation. Biol. Blood Marrow Transpl. 2008, 14, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Otabe, K.; Shimizu, N.; Komori, K.; Mizuno, M.; Katano, H.; Koga, H.; Sekiya, I. High-sensitivity virus and mycoplasma screening test reveals high prevalence of parvovirus B19 infection in human synovial tissues and bone marrow. Stem Cell Res. Ther. 2018, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Tada, K.; Chisaka, H.; Asao, H.; Sato, H.; Yaegashi, N.; Sugamura, K. Human parvovirus B19 induces cell cycle arrest at G(2) phase with accumulation of mitotic cyclins. J. Virol. 2001, 75, 7555–7563. [Google Scholar] [CrossRef]
- Komatsu, N.; Nakauchi, H.; Miwa, A.; Ishihara, T.; Eguchi, M.; Moroi, M.; Okada, M.; Sato, Y.; Wada, H.; Yawata, Y.; et al. Establishment and characterization of a human leukemic cell line with megakaryocytic features: Dependency on granulocyte-macrophage colony-stimulating factor, interleukin 3, or erythropoietin for growth and survival. Cancer Res. 1991, 51, 341–348. [Google Scholar]
- Manaresi, E.; Gallinella, G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses 2019, 11, 659. [Google Scholar] [CrossRef]
- Manaresi, E.; Bua, G.; Bonvicini, F.; Gallinella, G. A flow-FISH assay for the quantitative analysis of parvovirus B19 infected cells. J. Virol. Methods 2015, 223, 50–54. [Google Scholar] [CrossRef]
- Bonvicini, F.; Manaresi, E.; Bua, G.; Venturoli, S.; Gallinella, G. Keeping pace with parvovirus B19 genetic variability: A multiplex genotype-specific quantitative PCR assay. J. Clin. Microbiol. 2013, 51, 3753–3759. [Google Scholar] [CrossRef]
- Bonvicini, F.; Mirasoli, M.; Manaresi, E.; Bua, G.; Calabria, D.; Roda, A.; Gallinella, G. Single-cell chemiluminescence imaging of parvovirus B19 life cycle. Virus Res. 2013, 178, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, S.; Komatsu, N.; Frickhofen, N.; Anderson, S.; Kajigaya, S.; Young, N.S. First continuous propagation of B19 parvovirus in a cell line. Blood 1992, 79, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Goupille, O.; Penglong, T.; Lefevre, C.; Granger, M.; Kadri, Z.; Fucharoen, S.; Maouche-Chretien, L.; Leboulch, P.; Chretien, S. BET bromodomain inhibition rescues erythropoietin differentiation of human erythroleukemia cell line UT7. Biochem. Biophys. Res. Commun. 2012, 429, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chretien, S.; Varlet, P.; Verdier, F.; Gobert, S.; Cartron, J.P.; Gisselbrecht, S.; Mayeux, P.; Lacombe, C. Erythropoietin-induced erythroid differentiation of the human erythroleukemia cell line TF-1 correlates with impaired STAT5 activation. EMBO J. 1996, 15, 4174–4181. [Google Scholar] [CrossRef]
- Miyagawa, E.; Yoshida, T.; Takahashi, H.; Yamaguchi, K.; Nagano, T.; Kiriyama, Y.; Okochi, K.; Sato, H. Infection of the erythroid cell line, KU812Ep6 with human parvovirus B19 and its application to titration of B19 infectivity. J. Virol. Methods 1999, 83, 45–54. [Google Scholar] [CrossRef]
- Wong, S.; Keyvanfar, K.; Wan, Z.; Kajigaya, S.; Young, N.S.; Zhi, N. Establishment of an erythroid cell line from primary CD36+ erythroid progenitor cells. Exp. Hematol. 2010, 38, 994–1005. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Wong, S.; Heegaard, E.D.; Brown, K.E. Identification and characterization of a second novel human erythrovirus variant, A6. Virology 2002, 301, 374–380. [Google Scholar] [CrossRef]
- Mort, R.L.; Ford, M.J.; Sakaue-Sawano, A.; Lindstrom, N.O.; Casadio, A.; Douglas, A.T.; Keighren, M.A.; Hohenstein, P.; Miyawaki, A.; Jackson, I.J. Fucci2a: A bicistronic cell cycle reporter that allows Cre mediated tissue specific expression in mice. Cell Cycle 2014, 13, 2681–2696. [Google Scholar] [CrossRef] [PubMed]
- Bhukhai, K.; de Dreuzy, E.; Giorgi, M.; Colomb, C.; Negre, O.; Denaro, M.; Gillet-Legrand, B.; Cheuzeville, J.; Paulard, A.; Trebeden-Negre, H.; et al. Ex Vivo Selection of Transduced Hematopoietic Stem Cells for Gene Therapy of beta-Hemoglobinopathies. Mol. Ther. 2018, 26, 480–495. [Google Scholar] [CrossRef] [PubMed]
- Westerman, K.A.; Ao, Z.; Cohen, E.A.; Leboulch, P. Design of a trans protease lentiviral packaging system that produces high titer virus. Retrovirology 2007, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Bonsch, C.; Kempf, C.; Mueller, I.; Manning, L.; Laman, M.; Davis, T.M.; Ros, C. Chloroquine and its derivatives exacerbate B19V-associated anemia by promoting viral replication. PLoS Negl. Trop. Dis. 2010, 4, e669. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, P.P.; Humphries, R.K.; Moore, J.G.; Purcell, R.H.; Young, N.S. A human parvovirus-like virus inhibits haematopoietic colony formation in vitro. Nature 1983, 302, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Zhi, N.; Filippone, C.; Keyvanfar, K.; Kajigaya, S.; Brown, K.E.; Young, N.S. Ex vivo-generated CD36+ erythroid progenitors are highly permissive to human parvovirus B19 replication. J. Virol. 2008, 82, 2470–2476. [Google Scholar] [CrossRef] [PubMed]
- Bua, G.; Manaresi, E.; Bonvicini, F.; Gallinella, G. Parvovirus B19 Replication and Expression in Differentiating Erythroid Progenitor Cells. PLoS ONE 2016, 11, e0148547. [Google Scholar] [CrossRef]
- Kitamura, T.; Tange, T.; Terasawa, T.; Chiba, S.; Kuwaki, T.; Miyagawa, K.; Piao, Y.F.; Miyazono, K.; Urabe, A.; Takaku, F. Establishment and characterization of a unique human cell line that proliferates dependently on GM-CSF, IL-3, or erythropoietin. J. Cell Physiol. 1989, 140, 323–334. [Google Scholar] [CrossRef]
- Kitamura, T.; Tojo, A.; Kuwaki, T.; Chiba, S.; Miyazono, K.; Urabe, A.; Takaku, F. Identification and analysis of human erythropoietin receptors on a factor-dependent cell line, TF-1. Blood 1989, 73, 375–380. [Google Scholar] [CrossRef]
- Gallinella, G.; Manaresi, E.; Zuffi, E.; Venturoli, S.; Bonsi, L.; Bagnara, G.P.; Musiani, M.; Zerbini, M. Different patterns of restriction to B19 parvovirus replication in human blast cell lines. Virology 2000, 278, 361–367. [Google Scholar] [CrossRef]
- Ganaie, S.S.; Zou, W.; Xu, P.; Deng, X.; Kleiboeker, S.; Qiu, J. Phosphorylated STAT5 directly facilitates parvovirus B19 DNA replication in human erythroid progenitors through interaction with the MCM complex. PLoS Pathog. 2017, 13, e1006370. [Google Scholar] [CrossRef]
- Winkelmann, J.C.; Ward, J.; Mayeux, P.; Lacombe, C.; Schimmenti, L.; Jenkins, R.B. A translocated erythropoietin receptor gene in a human erythroleukemia cell line (TF-1) expresses an abnormal transcript and a truncated protein. Blood 1995, 85, 179–185. [Google Scholar] [CrossRef]
- Bonvicini, F.; Filippone, C.; Delbarba, S.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Parvovirus B19 genome as a single, two-state replicative and transcriptional unit. Virology 2006, 347, 447–454. [Google Scholar] [CrossRef]
- Bonvicini, F.; Filippone, C.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Functional analysis and quantitative determination of the expression profile of human parvovirus B19. Virology 2008, 381, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Zermati, Y.; Varet, B.; Hermine, O. TGF-beta1 drives and accelerates erythroid differentiation in the epo-dependent UT-7 cell line even in the absence of erythropoietin. Exp. Hematol. 2000, 28, 256–266. [Google Scholar] [CrossRef]
- Chretien, S.; Moreau-Gachelin, F.; Apiou, F.; Courtois, G.; Mayeux, P.; Dutrillaux, B.; Cartron, J.P.; Gisselbrecht, S.; Lacombe, C. Putative oncogenic role of the erythropoietin receptor in murine and human erythroleukemia cells. Blood 1994, 83, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Kadri, Z.; Shimizu, R.; Ohneda, O.; Maouche-Chretien, L.; Gisselbrecht, S.; Yamamoto, M.; Romeo, P.H.; Leboulch, P.; Chretien, S. Direct binding of pRb/E2F-2 to GATA-1 regulates maturation and terminal cell division during erythropoiesis. PLoS Biol 2009, 7, e1000123. [Google Scholar] [CrossRef] [PubMed]
- Pop, R.; Shearstone, J.R.; Shen, Q.; Liu, Y.; Hallstrom, K.; Koulnis, M.; Gribnau, J.; Socolovsky, M. A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef]
- Xu, P.; Zhou, Z.; Xiong, M.; Zou, W.; Deng, X.; Ganaie, S.S.; Kleiboeker, S.; Peng, J.; Liu, K.; Wang, S.; et al. Parvovirus B19 NS1 protein induces cell cycle arrest at G2-phase by activating the ATR-CDC25C-CDK1 pathway. PLoS Pathog. 2017, 13, e1006266. [Google Scholar] [CrossRef]
- Yunoki, M.; Tsujikawa, M.; Urayama, T.; Sasaki, Y.; Morita, M.; Tanaka, H.; Hattori, S.; Takechi, K.; Ikuta, K. Heat sensitivity of human parvovirus B19. Vox Sang. 2003, 84, 164–169. [Google Scholar] [CrossRef]
- Mani, B.; Gerber, M.; Lieby, P.; Boschetti, N.; Kempf, C.; Ros, C. Molecular mechanism underlying B19 virus inactivation and comparison to other parvoviruses. Transfusion 2007, 47, 1765–1774. [Google Scholar] [CrossRef]
- Blumel, J.; Schmidt, I.; Willkommen, H.; Lower, J. Inactivation of parvovirus B19 during pasteurization of human serum albumin. Transfusion 2002, 42, 1011–1018. [Google Scholar] [CrossRef]
- Hattori, S.; Yunoki, M.; Tsujikawa, M.; Urayama, T.; Tachibana, Y.; Yamamoto, I.; Yamamoto, S.; Ikuta, K. Variability of parvovirus B19 to inactivation by liquid heating in plasma products. Vox Sang. 2007, 92, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, N.; Niederhauser, I.; Kempf, C.; Stuhler, A.; Lower, J.; Blumel, J. Different susceptibility of B19 virus and mice minute virus to low pH treatment. Transfusion 2004, 44, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Crabol, Y.; Terrier, B.; Rozenberg, F.; Pestre, V.; Legendre, C.; Hermine, O.; Montagnier-Petrissans, C.; Guillevin, L.; Mouthon, L.; Groupe d’experts de l’Assistance Publique-Hopitaux de, P. Intravenous immunoglobulin therapy for pure red cell aplasia related to human parvovirus b19 infection: A retrospective study of 10 patients and review of the literature. Clin. Infect. Dis. 2013, 56, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Filippone, C.; Zhi, N.; Wong, S.; Lu, J.; Kajigaya, S.; Gallinella, G.; Kakkola, L.; Soderlund-Venermo, M.; Young, N.S.; Brown, K.E. VP1u phospholipase activity is critical for infectivity of full-length parvovirus B19 genomic clones. Virology 2008, 374, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Zhi, N.; Mills, I.P.; Lu, J.; Wong, S.; Filippone, C.; Brown, K.E. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J. Virol. 2006, 80, 5941–5950. [Google Scholar] [CrossRef]
- Wolfisberg, R.; Ruprecht, N.; Kempf, C.; Ros, C. Impaired genome encapsidation restricts the in vitro propagation of human parvovirus B19. J. Virol. Methods 2013, 193, 215–225. [Google Scholar] [CrossRef]
- Chen, A.Y.; Kleiboeker, S.; Qiu, J. Productive parvovirus B19 infection of primary human erythroid progenitor cells at hypoxia is regulated by STAT5A and MEK signaling but not HIFalpha. PLoS Pathog. 2011, 7, e1002088. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ducloux, C.; You, B.; Langelé, A.; Goupille, O.; Payen, E.; Chrétien, S.; Kadri, Z. Enhanced Cell-Based Detection of Parvovirus B19V Infectious Units According to Cell Cycle Status. Viruses 2020, 12, 1467. https://doi.org/10.3390/v12121467
Ducloux C, You B, Langelé A, Goupille O, Payen E, Chrétien S, Kadri Z. Enhanced Cell-Based Detection of Parvovirus B19V Infectious Units According to Cell Cycle Status. Viruses. 2020; 12(12):1467. https://doi.org/10.3390/v12121467
Chicago/Turabian StyleDucloux, Céline, Bruno You, Amandine Langelé, Olivier Goupille, Emmanuel Payen, Stany Chrétien, and Zahra Kadri. 2020. "Enhanced Cell-Based Detection of Parvovirus B19V Infectious Units According to Cell Cycle Status" Viruses 12, no. 12: 1467. https://doi.org/10.3390/v12121467
APA StyleDucloux, C., You, B., Langelé, A., Goupille, O., Payen, E., Chrétien, S., & Kadri, Z. (2020). Enhanced Cell-Based Detection of Parvovirus B19V Infectious Units According to Cell Cycle Status. Viruses, 12(12), 1467. https://doi.org/10.3390/v12121467