
Phylodynamic Analysis and Implication of HCV Genotype 4 Variability on Antiviral Drug Response and T-Cell Recognition



Abstract
1. Introduction
2. Materials and Methods
3. Results
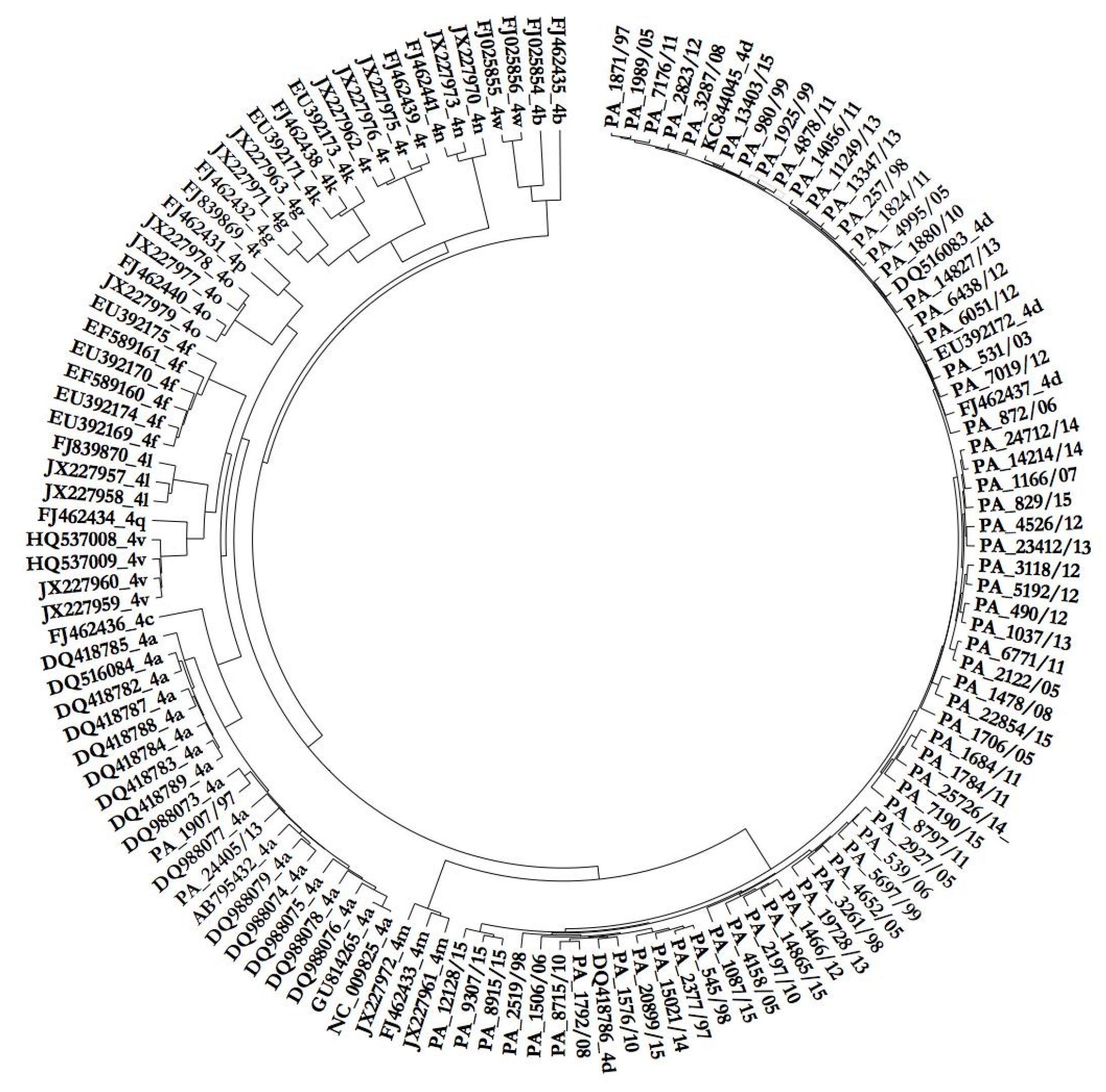
3.1. Phylogenetic Analysis of HCV Isolates
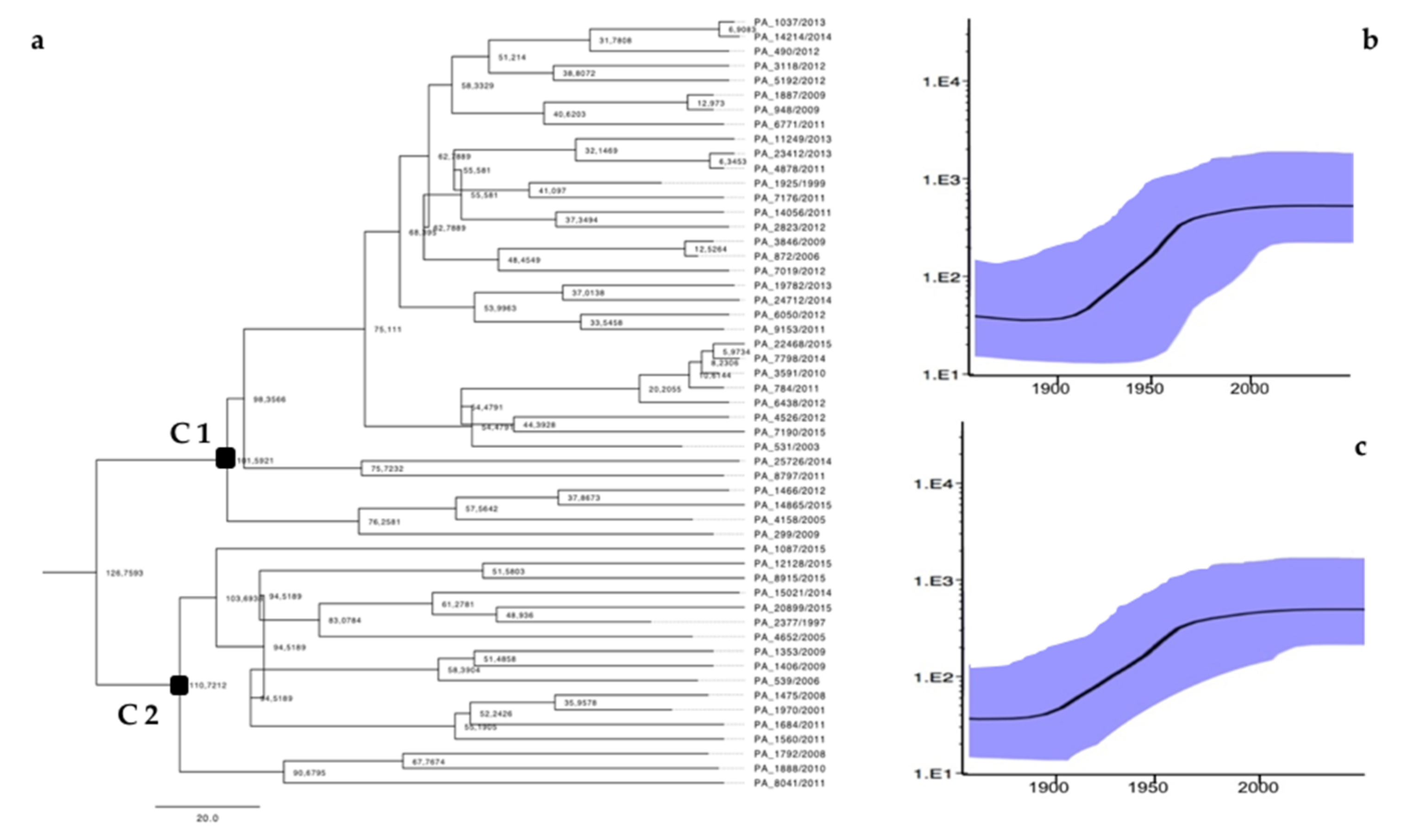
3.2. Phylodynamic Analysis
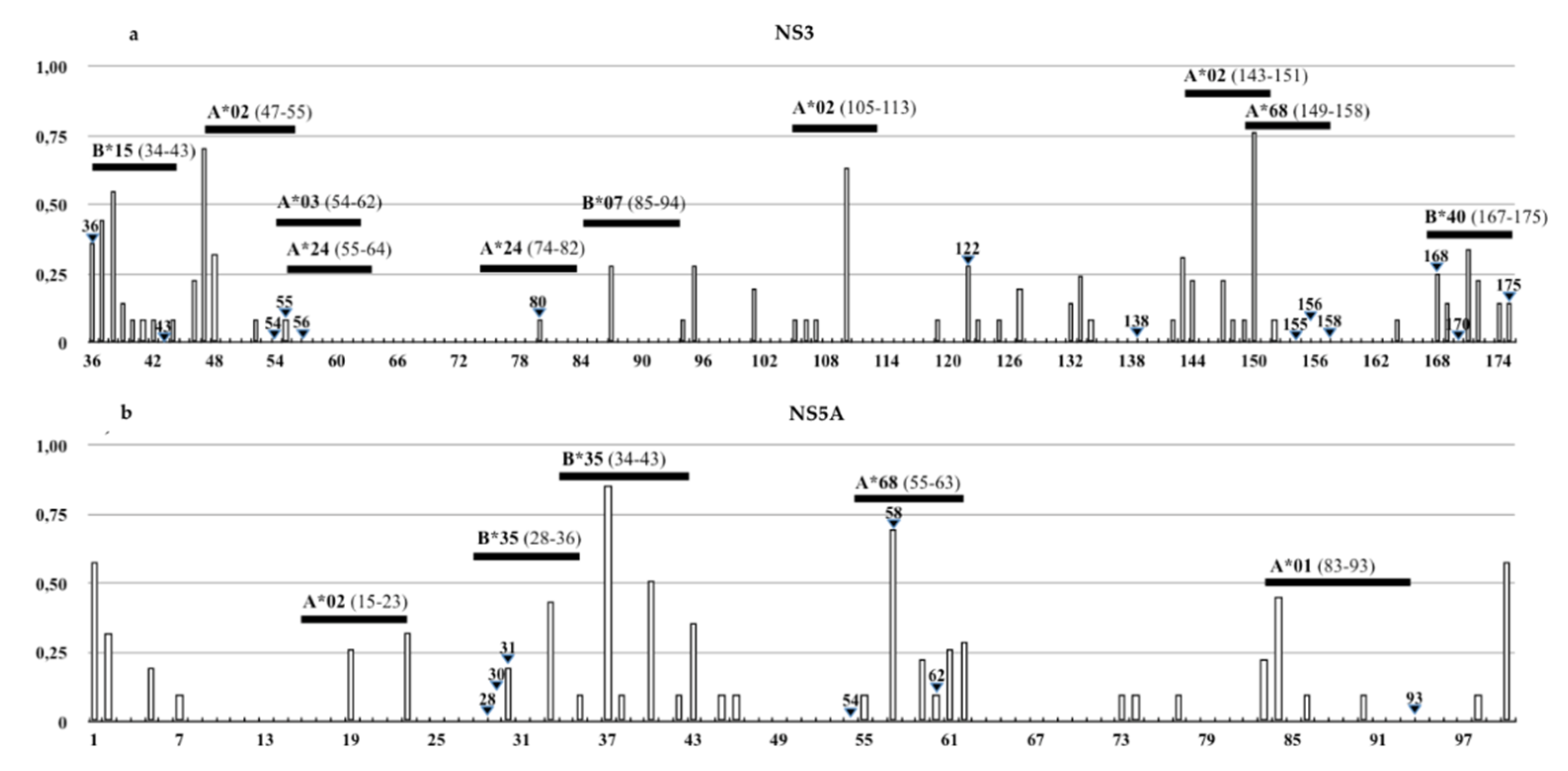
3.3. Identification of DAAs Resistance and Immune Escape Mutations
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ramia, S.; Eid-Fares, J. Distribution of hepatitis C virus genotypes in the Middle East. Int. J. Infect. Dis. 2006, 10, 272–277. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Messina, J.P.; Humphreys, I. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gower, E.; Estes, C. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Mohamed, M.K. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet 2000, 11, 887–891. [Google Scholar] [CrossRef]
- Payan, C.; Roudot-Thoraval, F. Changing of hepatitis C virus genotype patterns in France at the beginning of the third millenium: The GEMHEP GenoCII Study. J. Viral. Hepat. 2005, 12, 405–413. [Google Scholar] [CrossRef]
- Katsoulidou, A.; Sypsa, V. Molecular epidemiology of hepatitis C virus (HCV) in Greece: Temporal trends in HCV genotype-specific incidence and molecular characterization of genotype 4 isolates. J. Viral. Hepat. 2006, 13, 19–27. [Google Scholar] [CrossRef]
- Esteban, J.I.; Sauleda, S. The changing epidemiology of hepatitis C virus infection in Europe. J. Hepatol. 2008, 48, 148–162. [Google Scholar] [CrossRef]
- Ciccozzi, M.; Equestre, M. Hepatitis C virus genotype 4d in Southern Italy: Reconstruction of its origin and spread by a phylodynamic analysis. J. Med. Virol. 2012, 10, 1613–1619. [Google Scholar] [CrossRef]
- Pizzillo, P.; Almasio, P.L. HCV genotypes in Sicily: Is there any evidence of a shift? J. Med. Virol. 2009, 81, 1040–1046. [Google Scholar] [CrossRef]
- Ferraro, D.; Genovese, D. Phylogenetic reconstruction of HCV genotype 1b dissemination in a small city centre: The Camporeale model. J. Med. Virol. 2008, 80, 1723–1731. [Google Scholar] [CrossRef]
- EASL Guidelines. Available online: http://www.easl.eu (accessed on 28 November 2020).
- Gruener, N.H. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J. Virol. 2001, 75, 5550–5558. [Google Scholar] [CrossRef] [PubMed]
- Van Asten, L.; Verhaest, I. Spread of hepatitis C virus among European injection drug users infected with HIV: a phylogenetic analysis. J. Infect. Dis. 2004, 189, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214–218. [Google Scholar] [CrossRef] [PubMed]
- HCV Databases. Available online: http://hcv.lanl.gov (accessed on 28 November 2020).
- Datamonkey Adaptative Evolution Server. Available online: https://www.datamonkey.org/ (accessed on 28 November 2020).
- Genafor—Open Service for Medical Research. Available online: http://hcv.geno2pheno.org/index.php (accessed on 28 November 2020).
- Lontok, E.; Harrington, P. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 5, 1623–1632. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 1, 70–86. [Google Scholar] [CrossRef]
- Fevery, B.; Verbinnen, T. Virology analyses of HCV genotype 4 isolates from patients treated with simeprevir and peginterferon/ribavirin in the Phase III RESTORE study. J. Viral. Hepat. 2017, 24, 28–36. [Google Scholar] [CrossRef]
- Bartolini, B.; Lionetti, R. Dynamics of HCV genotype 4 resistance-associated variants during virologic escape with pIFN/RBV+daclatasvir: A case study using ultra deep pyrosequencing. J. Clin. Virol. 2015, 66, 38–43. [Google Scholar] [CrossRef]
- Van de Vijver, D.A.; Wensing, A.M. The calculated genetic barrier for antiretroviral drug resistance substitutions is largely similar for different HIV-1 subtypes. J. Acquir. Immune. Defic. Syndr. 2006, 41, 352–360. [Google Scholar] [CrossRef]
- Ferraro, D.; Urone, N. HCV-1b intra-subtype variability: Impact on genetic barrier to protease inhibitors. Infect. Genet. Evol. 2014, 23, 80–85. [Google Scholar] [CrossRef]
- Free Epitopes Database and Prediction Resources. Available online: https://www.iedb.org/ (accessed on 28 November 2020).
- Allele Frequency Net Database. Available online: http://www.allelefrequencies.net/ (accessed on 28 November 2020).
- Nielsen, M.; Lundegaard, C. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein. Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Schnell, G.; Tripathi, R. Hepatitis C virus genotype 4 resistance and subtype demographic characterization of patients treated with ombitasvir plus paritaprevir/ritonavir. Antimicrob. Agents Chemother. 2015, 59, 6807–6815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marascio, N.; Liberto, M. Update on epidemiology of HCV in Italy: Focus on the Calabria Region. BMC Infect. Dis. 2014, 14, 1471–2334. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Cochrane, A. The hepatitis C virus epidemic among injecting drug users. Infect. Genet. Evol. 2005, 2, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Rajhi, M.; Ghedira, K. Phylogenetic Analysis and Epidemic History of Hepatitis C Virus Genotype 2 in Tunisia, North Africa. PLoS ONE 2016, 11, e0153761. [Google Scholar] [CrossRef]
- Zhou, N.; Hernandez, D. NS5A Sequence Heterogeneity and Mechanisms of Daclatasvir Resistance in Hepatitis C Virus Genotype 4 Infection. J. Infect. Dis. 2016, 213, 206–215. [Google Scholar] [CrossRef][Green Version]
- Bell, A.M.; Wagner, J.L. Elbasvir/Grazoprevir: A Review of the Latest Agent in the Fight against Hepatitis C. Int. J. Hepatol. 2016, 2016, 3852126. [Google Scholar] [CrossRef]
- Vijgen, L.; Verbeeck, J. A cellular replicon-based phenotyping assay to determine susceptibility of hepatitis C virus clinical isolates to NS3/4A protease inhibitors. Methods. Mol. Biol. 2013, 2, 37–41. [Google Scholar]
- Bin, H.; Bandita, P. In Vitro Susceptibility of Hepatitis C Virus Genotype 1 through 6 Clinical Isolates to the Pangenotypic NS3/4A Inhibitor Voxilaprevir. J. Clin. Microbiol. 2020, 57, 173–177. [Google Scholar]
- Halfon, P.; Locarnini, S. Hepatitis C virus resistance to protease inhibitors. J. Hepatol. 2011, 55, 192–206. [Google Scholar] [CrossRef]
- Pawlotsky, J. Retreatment of Hepatitis C Virus-Infected Patients with Direct-Acting Antiviral Failures. Int. J. Hepatol. 2016, 38, 21–26. [Google Scholar] [CrossRef]
- Computational analysis of naturally occurring resistance-associated substitutions in genes NS3, NS5A, and NS5B among 86 subtypes of hepatitis C virus worldwide. Infect. Drug. Resist. 2019, 8, 17–23.
- Gaudieri, S.; Rauch, A. Hepatitis C virus drug resistance and immune-driven adaptations: Relevance to new antiviral therapy. Hepatology 2009, 49, 1069–1082. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Isolates | Cluster | HIV Co-Infection | Age (Years) | Route of Transmissions | |||||
|---|---|---|---|---|---|---|---|---|---|
| <30 | 30–50 | >50 | S | B | DU | U | |||
| 36 | C1 | 11 (30.5%) | 4 (11%) | 22 (61%) | 10 (27%) | 3 (0.8%) | 2 (0.5%) | 14 (39%) | 17 (47%) |
| 17 | C2 | 0 | 0 | 4 (11.7%) | 13 (85%) | 0 | 10 (58%) | 0 | 7 (42%) |
| RAS NS3 | Frequency | Resistance to | RAS NS5A | Frequency | Resistance to |
|---|---|---|---|---|---|
| 36L | 92% | GZP, SMP | 30R | 100% | DCV |
| 122T | 92% | SMP | 58T | 22% | DCV |
| 122A | 8% | SMP/PRV | 62Q | 5% | DCV |
| 36C | 6% | GZP | 62A | 2% | DCV |
| 36A | 2% | GZP | 31V | 2% | DCV, LDV |
| 80K | 2% | VXP | 58S | 2% | DCV |
| 168E | 2% | VXP | |||
| 168N | 2% | VXP/PRV |
| Epitope Position (Gene/aa.) | HLA-I Allele | IEDB Epitope Sequences | Mutations in Sicilian HCV-4d Strain | Change on HLA-Binding Affinity |
|---|---|---|---|---|
| NS3/54-62 | A*03:01 | TVYHGAGTK | V55A | High to intermediate |
| NS3/55-64 | A*24:02 | VYHGAGSKTL | V55A | Intermediate to low |
| NS3/74-82 | A*24:02 | MYTNVDQDL | Q80K | Lack of binding |
| NS3/167-175 | B*40:02 | VDFVPVESM | D168N | Lack of binding |
| NS5A/34-43 | B*35:01 | VPFFSCQRGY | F38L | Lack of binding |
| NS5A/55-63 | A*68:02 | TTCPCGAQI | T56R | Lack of binding |
| NS5A/83-93 | A*01:01 | MWSGTFPINAY | M83T, W84C, S85H/N/R | Lack of binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colomba, G.M.E.; Urone, N.; Marco, V.d.; Ferraro, D. Phylodynamic Analysis and Implication of HCV Genotype 4 Variability on Antiviral Drug Response and T-Cell Recognition. Viruses 2020, 12, 1363. https://doi.org/10.3390/v12121363
Colomba GME, Urone N, Marco Vd, Ferraro D. Phylodynamic Analysis and Implication of HCV Genotype 4 Variability on Antiviral Drug Response and T-Cell Recognition. Viruses. 2020; 12(12):1363. https://doi.org/10.3390/v12121363
Chicago/Turabian StyleColomba, Giuseppina Maria Elena, Noemi Urone, Vito di Marco, and Donatella Ferraro. 2020. "Phylodynamic Analysis and Implication of HCV Genotype 4 Variability on Antiviral Drug Response and T-Cell Recognition" Viruses 12, no. 12: 1363. https://doi.org/10.3390/v12121363
APA StyleColomba, G. M. E., Urone, N., Marco, V. d., & Ferraro, D. (2020). Phylodynamic Analysis and Implication of HCV Genotype 4 Variability on Antiviral Drug Response and T-Cell Recognition. Viruses, 12(12), 1363. https://doi.org/10.3390/v12121363