
Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses
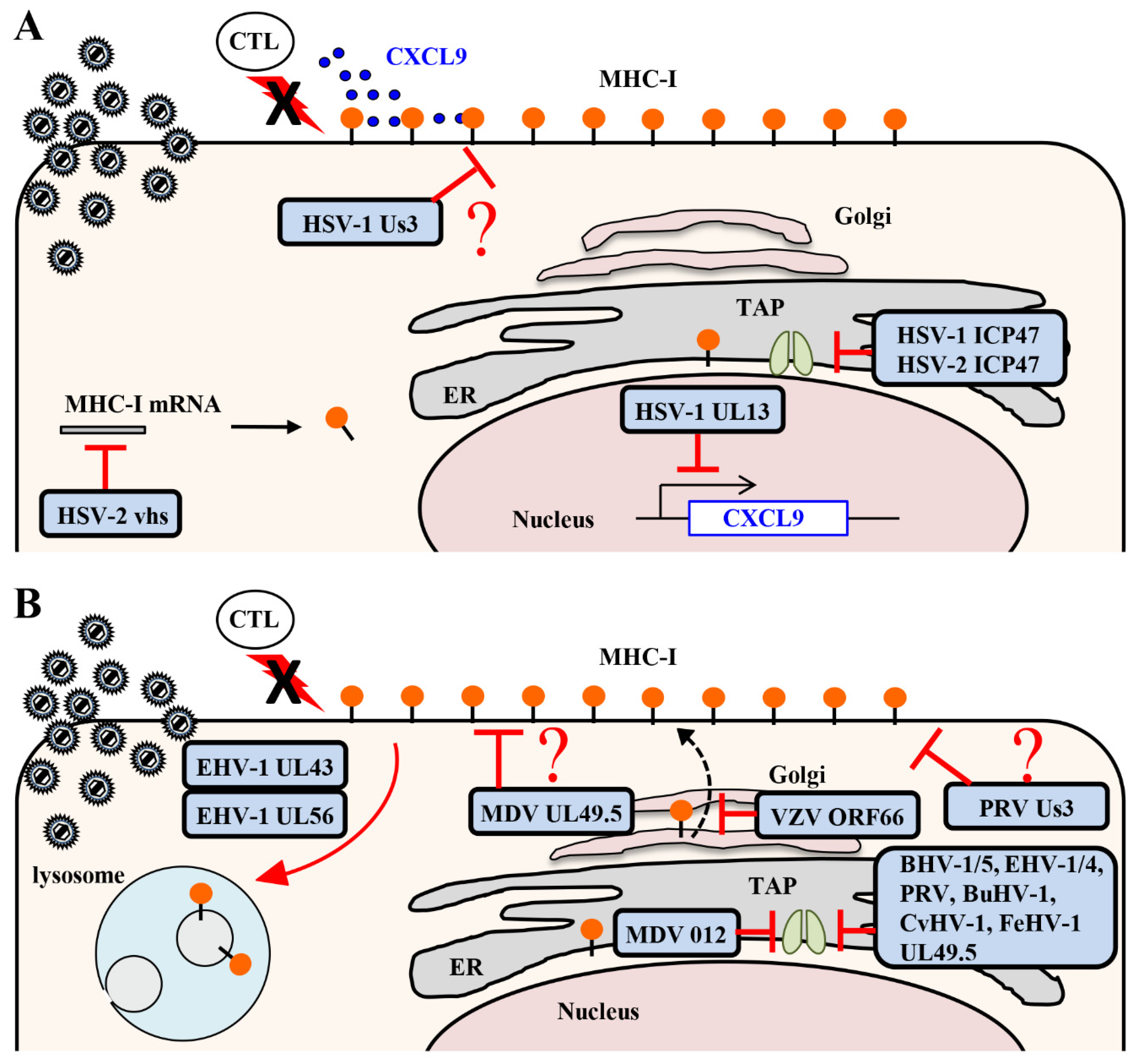
2.1. MHC Class I Antigen Presentation to CTLs
2.1.1. Inhibition of MHC Class I Antigen Presentation by HSV
2.1.2. Inhibition of MHC Class I Antigen Presentation by Varicelloviruses
2.1.3. Inhibition of MHC Class I Antigen Presentation by Mardiviruses
2.2. Infiltration of CTLs at Infection Sites by Chemokines
Downregulation of Chemokine CXCL9 Expression by HSV-1
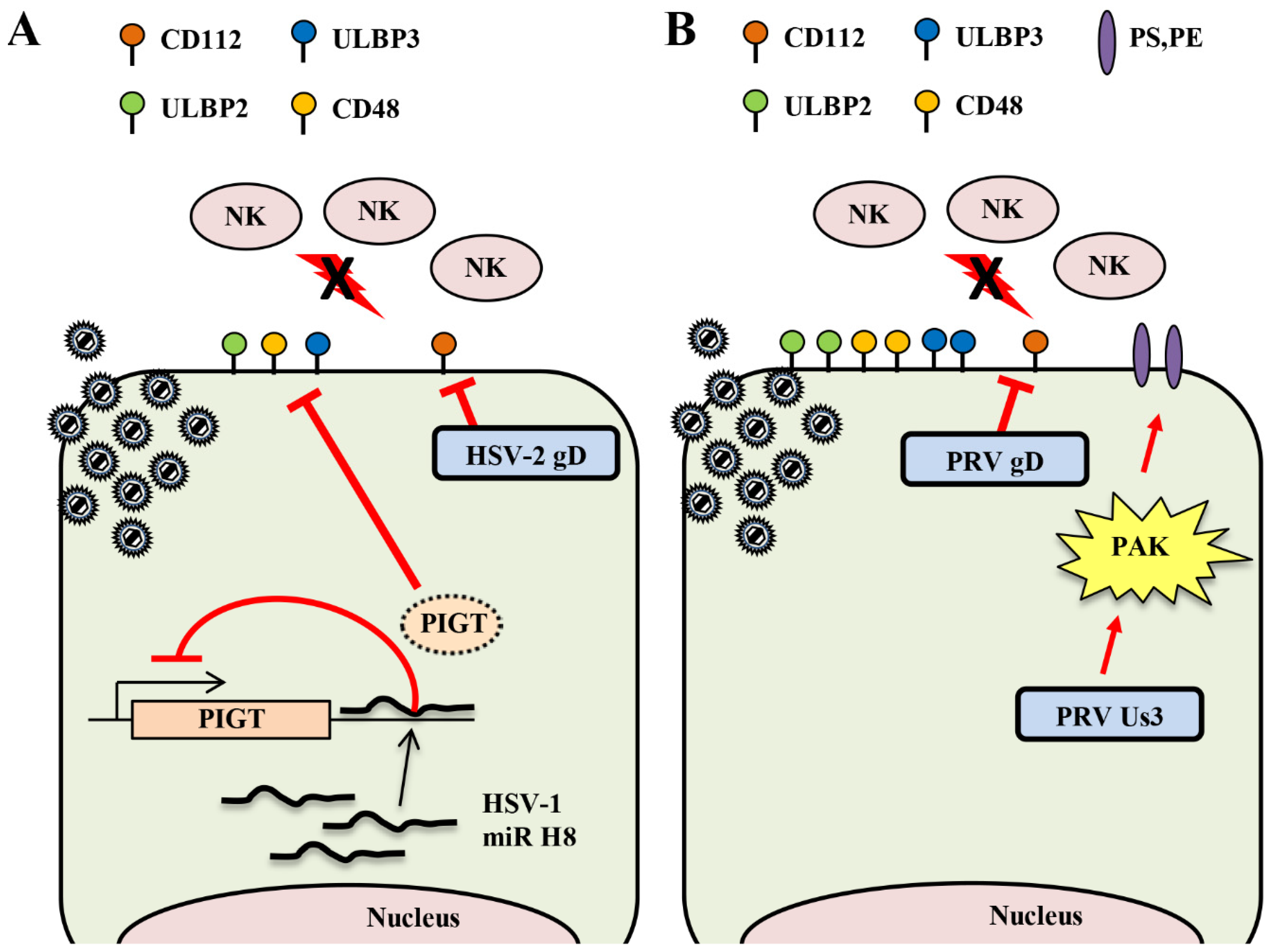
2.3. Natural Killer Cells
2.3.1. Downregulation of the Cell Surface Expression of Activating Ligands by HSV
2.3.2. Downregulation of the Surface Expression of NK Activating Ligands by Varicelloviruses
2.3.3. Upregulation of the Surface Expression of NK Inhibitory Ligands by Varicelloviruses
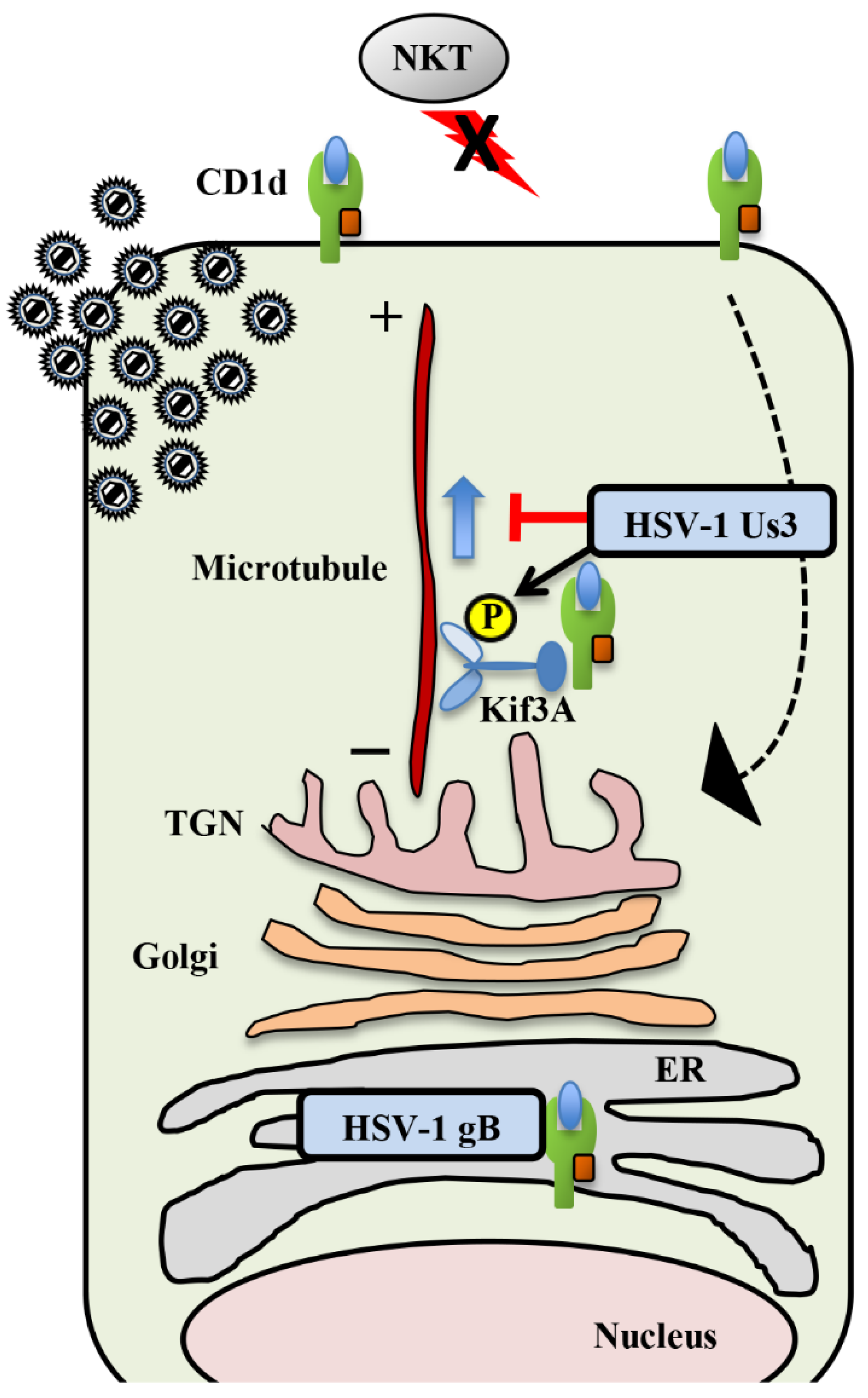
2.4. Natural Killer T Cells
Inhibition of CD1d Recycling by HSV-1 Us3 and gB
3. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular Biology of Pseudorabies Virus: Impact on Neurovirology and Veterinary Medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Azab, W.; Osterrieder, N. Equine herpesviruses type 1 (EHV-1) and 4 (EHV-4)—Masters of co-evolution and a constant threat to equids and beyond. Vet. Microbiol. 2013, 167, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Tischer, B.K.; Trapp, S.; Osterrieder, N. Marek’s disease virus: Lytic replication, oncogenesis and control. Expert Rev. Vaccines 2006, 5, 761–772. [Google Scholar] [CrossRef]
- Woźniakowski, G.; Samorek-Salamonowicz, E. Animal herpesviruses and their zoonotic potential for cross-species infection. Ann. Agric. Environ. Med. 2015, 22, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef]
- Luecke, S.; Paludan, S.R. Innate recognition of alphaherpesvirus DNA. Adv. Virus Res. 2015, 92, 63–100. [Google Scholar]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, C. A Tug of War: DNA-Sensing Antiviral Innate Immunity and Herpes Simplex Virus Type I Infection. Front. Microbiol. 2019, 10, 2627. [Google Scholar] [CrossRef]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef]
- Horst, D.; Verweij, M.C.; Davison, A.J.; Ressing, M.E.; Wiertz, E.J. Viral evasion of T cell immunity: Ancient mechanisms offering new applications. Curr. Opin. Immunol. 2011, 23, 96–103. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Krmpotic, A.; Messerle, M.; Crnkovic-Mertens, I.; Polic, B.; Jonjic, S.; Koszinowski, U.H. The Immunoevasive Function Encoded by the Mouse Cytomegalovirus Gene m152 Protects the Virus against T Cell Control in Vivo. J. Exp. Med. 1999, 190, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.G.; May, J.S.; Smith, X.; Marques, S.; Adler, H.S.; Koszinowski, U.H.; Simas, J.; Efstathiou, S. K3-mediated evasion of CD8+ T cells aids amplification of a latent γ-herpesvirus. Nat. Immunol. 2002, 3, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Jugovic, P.; Hill, A.M.; Tomazin, R.; Ploegh, H.; Johnson, D.C. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. J. Virol. 1998, 72, 5076–5084. [Google Scholar] [CrossRef]
- Hill, A.; Jugovic, P.; York, I.; Russ, G.; Bennink, J.; Yewdell, J.; Ploegh, H.; Johnson, D. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995, 375, 411–415. [Google Scholar] [CrossRef]
- Früh, K.; Ahn, K.; Djaballah, H.; Sempé, P.; Van Endert, P.M.; Tampé, R.; Peterson, P.A.; Yang, Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375, 415–418. [Google Scholar] [CrossRef]
- Oldham, M.L.; Hite, R.K.; Steffen, A.M.; Damko, E.; Li, Z.; Walz, T.; Chen, J. A mechanism of viral immune evasion revealed by cryo-EM analysis of the TAP transporter. Nature 2016, 529, 537–540. [Google Scholar] [CrossRef]
- Oldham, M.L.; Grigorieff, N.; Chen, J. Structure of the transporter associated with antigen processing trapped by herpes simplex virus. eLife 2016, 5, e21829. [Google Scholar] [CrossRef]
- Ahn, K.; Meyer, T.H.; Uebel, S.; Sempe, P.; Djaballah, H.; Yang, Y.; Peterson, P.A.; Fruh, K.; Tampe, R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996, 15, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected Cell Protein (ICP)47 Enhances Herpes Simplex Virus Neurovirulence by Blocking the CD8+ T Cell Response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Arii, J.; Shiratori, I.; Akashi, H.; Arase, H.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 2009, 83, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Koyanagi, N.; Ogawa, R.; Shindo, K.; Suenaga, T.; Sato, A.; Arii, J.; Kato, A.; Kiyono, H.; Arase, H.; et al. Us3 Kinase Encoded by Herpes Simplex Virus 1 Mediates Downregulation of Cell Surface Major Histocompatibility Complex Class I and Evasion of CD8+ T Cells. PLoS ONE 2013, 8, e72050. [Google Scholar] [CrossRef]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef]
- Smiley, J.R. Herpes Simplex Virus Virion Host Shutoff Protein: Immune Evasion Mediated by a Viral RNase? J. Virol. 2004, 78, 1063–1068. [Google Scholar] [CrossRef]
- Read, G.S. Virus-encoded endonucleases: Expected and novel functions. Wiley Interdiscip. Rev. RNA 2013, 4, 693–708. [Google Scholar] [CrossRef]
- Tigges, M.A.; Leng, S.; Johnson, D.C.; Burke, R.L. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J. Immunol. 1996, 156, 3901–3910. [Google Scholar]
- Murphy, J.A.; Duerst, R.J.; Smith, T.J.; Morrison, L.A. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 2003, 77, 9337–9345. [Google Scholar] [CrossRef]
- Abendroth, A.; Lin, I.; Slobedman, B.; Ploegh, H.; Arvin, A.M. Varicella-Zoster Virus Retains Major Histocompatibility Complex Class I Proteins in the Golgi Compartment of Infected Cells. J. Virol. 2001, 75, 4878–4888. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Yee, M.B.; Erazo, A.; Abendroth, A.; Kinchington, P.R. Downregulation of Class I Major Histocompatibility Complex Surface Expression by Varicella-Zoster Virus Involves Open Reading Frame 66 Protein Kinase-Dependent and -Independent Mechanisms. J. Virol. 2007, 81, 9034–9049. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deruelle, M.J.; Broeke, C.V.D.; Nauwynck, H.J.; Mettenleiter, T.C.; Favoreel, H.W. Pseudorabies virus US3- and UL49.5-dependent and -independent downregulation of MHC I cell surface expression in different cell types. Virology 2009, 395, 172–181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- Koppers-Lalic, D.; Reitst, E.A.J.; Ressing, M.E.; Lipinska, A.D.; Abele, R.; Koch, J.; Rezende, M.M.; Admiraal, P.; Van Leeuwen, D.; Bienkowska-Szewczyk, K.; et al. Varicelloviruses avoid T cell recognition by UL49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 2005, 102, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Verweij, M.C.; Lipińska, A.D.; Wang, Y.; Quinten, E.; Reits, E.A.; Koch, J.; Loch, S.; Rezende, M.M.; Daus, F.; et al. Varicellovirus UL49.5 Proteins Differentially Affect the Function of the Transporter Associated with Antigen Processing, TAP. PLoS Pathog. 2008, 4, e1000080. [Google Scholar] [CrossRef]
- Verweij, M.C.; Lipińska, A.; Koppers-Lalic, D.; Van Leeuwen, W.F.; Cohen, J.I.; Kinchington, P.R.; Messaoudi, I.; Bieńkowska-Szewczyk, K.; Ressing, M.E.; Rijsewijk, F.A.M.; et al. The Capacity of UL49.5 Proteins To Inhibit TAP Is Widely Distributed among Members of the Genus Varicellovirus. J. Virol. 2011, 85, 2351–2363. [Google Scholar] [CrossRef]
- Said, A.; Azab, W.; Damiani, A.; Osterrieder, N. Equine Herpesvirus Type 4 UL56 and UL49.5 Proteins Downregulate Cell Surface Major Histocompatibility Complex Class I Expression Independently of Each Other. J. Virol. 2012, 86, 8059–8071. [Google Scholar] [CrossRef]
- Verweij, M.C.; Koppers-Lalic, D.; Loch, S.; Klauschies, F.; De La Salle, H.; Quinten, E.; Lehner, P.J.; Mulder, A.; Knittler, M.R.; Tampé, R.; et al. The varicellovirus UL49.5 protein blocks the transporter associated with antigen processing (TAP) by inhibiting essential conformational transitions in the 6+6 transmembrane TAP core complex. J. Immunol. 2008, 181, 4894–4907. [Google Scholar] [CrossRef]
- Osterrieder, N.; Feineis, S.; Osterrieder, N.; Van De Walle, G.R. Identification and Characterization of Equine Herpesvirus Type 1 pUL56 and Its Role in Virus-Induced Downregulation of Major Histocompatibility Complex Class I. J. Virol. 2012, 86, 3554–3563. [Google Scholar] [CrossRef][Green Version]
- Huang, T.; Lehmann, M.J.; Said, A.; Ma, G.; Osterrieder, N. Major Histocompatibility Complex Class I Downregulation Induced by Equine Herpesvirus Type 1 pUL56 Is through Dynamin-Dependent Endocytosis. J. Virol. 2014, 88, 12802–12815. [Google Scholar] [CrossRef]
- Huang, T.; Ma, G.; Osterrieder, N. Equine Herpesvirus 1 Multiply Inserted Transmembrane Protein pUL43 Cooperates with pUL56 in Downregulation of Cell Surface Major Histocompatibility Complex Class I. J. Virol. 2015, 89, 6251–6263. [Google Scholar] [CrossRef]
- Gimeno, I.M.; Witter, R.L.; Hunt, H.D.; Lee, L.F.; Reddy, S.M.; Neumann, U. Marek’s Disease Virus Infection in the Brain: Virus Replication, Cellular Infiltration, and Major Histocompatibility Complex Antigen Expression. Vet. Pathol. 2001, 38, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Hunt, H.; Lupiani, B.; Miller, M.; Gimeno, I.; Lee, L.; Parcells, M. Marek’s Disease Virus Down-Regulates Surface Expression of MHC (B Complex) Class I (BF) Glycoproteins during Active but not Latent Infection of Chicken Cells. Virology 2001, 282, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Jarosinski, K.W.; Hunt, H.D.; Osterrieder, N. Down-regulation of MHC class I by the Marek’s disease virus (MDV) UL49.5 gene product mildly affects virulence in a haplotype-specific fashion. Virology 2010, 405, 457–463. [Google Scholar] [CrossRef]
- Hearn, C.; Preeyanon, L.; Hunt, H.D.; York, I.A. An MHC class I immune evasion gene of Marek’s disease virus. Virology 2015, 475, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Thapa, M.; Carr, D.J. Chemokines and Chemokine Receptors Critical to Host Resistance Following Genital Herpes Simplex Virus Type 2 (HSV-2) Infection. Open Immunol. J. 2008, 1, 33–41. [Google Scholar] [CrossRef][Green Version]
- Todd, R.W. The role of chemokines during herpes simplex virus-1 infection. Front. Biosci. 2008, 13, 4862–4872. [Google Scholar] [CrossRef][Green Version]
- Melchjorsen, J.; Sørensen, L.N.; Rasmussen, S.B. Expression and function of chemokines during viral infections: From molecular mechanisms to in vivo function. J. Leukoc. Biol. 2003, 74, 331–343. [Google Scholar] [CrossRef]
- Thapa, M.; Welner, R.S.; Pelayo, R.; Carr, D.J.J. CXCL9 and CXCL10 Expression Are Critical for Control of Genital Herpes Simplex Virus Type 2 Infection through Mobilization of HSV-Specific CTL and NK Cells to the Nervous System. J. Immunol. 2008, 180, 1098–1106. [Google Scholar] [CrossRef]
- Zhang, M.; Deng, X.; Guan, X.; Geng, L.; Fu, M.; Zhang, B.; Chen, R.; Hu, H.; Hu, K.; Zhang, D.; et al. Herpes Simplex Virus Type 2 Infection-Induced Expression of CXCR3 Ligands Promotes CD4+ T Cell Migration and Is Regulated by the Viral Immediate-Early Protein ICP4. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Shin, H.; Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012, 491, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A. Viral mimicry of cytokines, chemokines and their receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Pontejo, S.M.; Murphy, P.M.; Pease, J.E. Chemokine Subversion by Human Herpesviruses. J. Innate Immun. 2018, 10, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Coulter, L.J.; Moss, H.W.M.; Lang, J.; McGeoch, D.J. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol. 1993, 74 Pt 3, 387–395. [Google Scholar] [CrossRef]
- Purves, F.C.; Ogle, W.O.; Roizman, B. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. USA 1993, 90, 6701–6705. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kato, K.; Tanaka, M.; Kanamori, M.; Nishiyama, Y.; Yamanashi, Y. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 2003, 77, 2359–2368. [Google Scholar] [CrossRef]
- Koyanagi, N.; Imai, T.; Shindo, K.; Satoshi, U.; Fujii, W.; Ichinohe, T.; Takemura, N.; Kakuta, S.; Uematsu, S.; Kiyono, H.; et al. Herpes simplex virus-1 evasion of CD8+ T cell accumulation contributes to viral encephalitis. J. Clin. Investig. 2017, 127, 3784–3795. [Google Scholar] [CrossRef]
- Lanier, L.L. Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 2008, 8, 259–268. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.S.; Geraghty, R.J.; Martinez, W.M.; Montgomery, R.I.; Whitbeckbcd, J.C.; Xu, R.; Eisenbergcd, R.J.; Cohenbc, G.H.; Spear, P.G. A Cell Surface Protein with Herpesvirus Entry Activity (HveB) Confers Susceptibility to Infection by Mutants of Herpes Simplex Virus Type 1, Herpes Simplex Virus Type 2, and Pseudorabies Virus. Virology 1998, 246, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Krummenacher, C.; Baribaud, F.; De Leon, M.P.; Baribaud, I.; Whitbeck, J.; Xu, R.; Cohen, G.H.; Eisenberg, R.J. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 2004, 322, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Stiles, K.M.; Milne, R.S.B.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. The herpes simplex virus receptor nectin-1 is down-regulated after trans-interaction with glycoprotein D. Virology 2008, 373, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Grauwet, K.; Cantoni, C.; Parodi, M.; De Maria, A.; Devriendt, B.; Pende, D.; Moretta, L.; Vitale, M.; Favoreel, H.W. Modulation of CD112 by the alphaherpesvirus gD protein suppresses DNAM-1-dependent NK cell-mediated lysis of infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 16118–16123. [Google Scholar] [CrossRef]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Levi, A.; Weisblum, Y.; Yamin, R.; Charpak-Amikam, Y.; Wolf, D.G.; Mandelboim, O. HSV1 MicroRNA Modulation of GPI Anchoring and Downstream Immune Evasion. Cell Rep. 2016, 17, 949–956. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-anchored proteins: Special emphasis on GPI lipid remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef]
- Lankry, D.; Simic, H.; Klieger, Y.; Levi-Schaffer, F.; Jonjic, S.; Mandelboim, O. Expression and Function of CD300 in NK Cells. J. Immunol. 2010, 185, 2877–2886. [Google Scholar] [CrossRef]
- Lankry, D.; Rovis, T.L.; Jonjic, S.; Mandelboim, O. The interaction between CD 300a and phosphatidylserine inhibits tumor cell killing by NK cells. Eur. J. Immunol. 2013, 43, 2151–2161. [Google Scholar] [CrossRef]
- Grauwet, K.; Vitale, M.; De Pelsmaeker, S.; Jacob, T.; Laval, K.; Moretta, L.; Parodi, M.; Parolini, S.; Cantoni, C.; Favoreel, H.W. Pseudorabies Virus US3 Protein Kinase Protects Infected Cells from NK Cell-Mediated Lysis via Increased Binding of the Inhibitory NK Cell Receptor CD300a. J. Virol. 2016, 90, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E.; Baker, S.M.; Loren, C.P.; Haley, K.M.; Itakura, A.; Pang, J.; Greenberg, D.L.; David, L.L.; Manser, E.; Chernoff, J.; et al. The PAK system links Rho GTPase signaling to thrombin-mediated platelet activation. Am. J. Physiol. Cell Physiol. 2013, 305, C519–C528. [Google Scholar] [CrossRef]
- Broeke, C.V.D.; Radu, M.; Deruelle, M.; Nauwynck, H.; Hofmann, C.; Jaffer, Z.M.; Chernoff, J.; Favoreel, H.W. Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. USA 2009, 106, 8707–8712. [Google Scholar] [CrossRef] [PubMed]
- Bendelac, A.; Savage, P.B.; Teyton, L. The Biology of NKT Cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [PubMed]
- Grubor-Bauk, B.; Simmons, A.; Mayrhofer, G.; Speck, P.G. Impaired clearance of herpes simplex virus type 1 from mice lacking CD1d or NKT cells expressing the semivariant V alpha 14-J alpha 281 TCR. J. Immunol. 2003, 170, 1430–1434. [Google Scholar] [CrossRef]
- Grubor-Bauk, B.; Arthur, J.L.; Mayrhofer, G. Importance of NKT Cells in Resistance to Herpes Simplex Virus, Fate of Virus-Infected Neurons, and Level of Latency in Mice. J. Virol. 2008, 82, 11073–11083. [Google Scholar] [CrossRef]
- Cornish, A.L.; Keating, R.; Kyparissoudis, K.; Smyth, M.J.; Carbone, F.R.; Godfrey, D.I. NKT cells are not critical for HSV-1 disease resolution. Immunol. Cell Biol. 2006, 84, 13–19. [Google Scholar] [CrossRef]
- Yuan, W.; Dasgupta, A.; Cresswell, P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat. Immunol. 2006, 7, 835–842. [Google Scholar] [CrossRef]
- Rao, V.; Pham, H.T.; Kulkarni, A.; Yang, Y.; Liu, X.; Knipe, D.M.; Cresswell, P.; Yuan, W. Herpes Simplex Virus 1 Glycoprotein B and US3 Collaborate to Inhibit CD1d Antigen Presentation and NKT Cell Function. J. Virol. 2011, 85, 8093–8104. [Google Scholar] [CrossRef]
- Xiong, R.; Rao, P.; Kim, S.; Li, M.; Wen, X.; Yuan, W. Herpes Simplex Virus 1 US3 Phosphorylates Cellular KIF3A to Downregulate CD1d Expression. J. Virol. 2015, 89, 6646–6655. [Google Scholar] [CrossRef]
- Rao, P.; Wen, X.; Lo, J.H.; Kim, S.; Li, X.; Chen, S.; Feng, X.; Akbari, O.; Yuan, W. Herpes Simplex Virus 1 Specifically Targets Human CD1d Antigen Presentation to Enhance Its Pathogenicity. J. Virol. 2018, 92, 22. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.M.; McSharry, B.P.; Steain, M.; Slobedman, B.; Abendroth, A. Varicella-Zoster Virus and Herpes Simplex Virus 1 Differentially Modulate NKG2D Ligand Expression during Productive Infection. J. Virol. 2015, 89, 7932–7943. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koyanagi, N.; Kawaguchi, Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses 2020, 12, 1354. https://doi.org/10.3390/v12121354
Koyanagi N, Kawaguchi Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses. 2020; 12(12):1354. https://doi.org/10.3390/v12121354
Chicago/Turabian StyleKoyanagi, Naoto, and Yasushi Kawaguchi. 2020. "Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses" Viruses 12, no. 12: 1354. https://doi.org/10.3390/v12121354
APA StyleKoyanagi, N., & Kawaguchi, Y. (2020). Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses, 12(12), 1354. https://doi.org/10.3390/v12121354