Characterization of the Vaginal DNA Virome in Health and Dysbiosis

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Characterisation of the Bacterial Component of the VMB
2.3. Virus Like Particles (VLP) Purification, Viral DNA Extraction, and Sequencing
2.4. Viral-Operational Taxonomic Unit (vOTU) Table
2.5. Community Analysis
2.6. Ethics Statement
3. Results
3.1. Samples and Sequencing
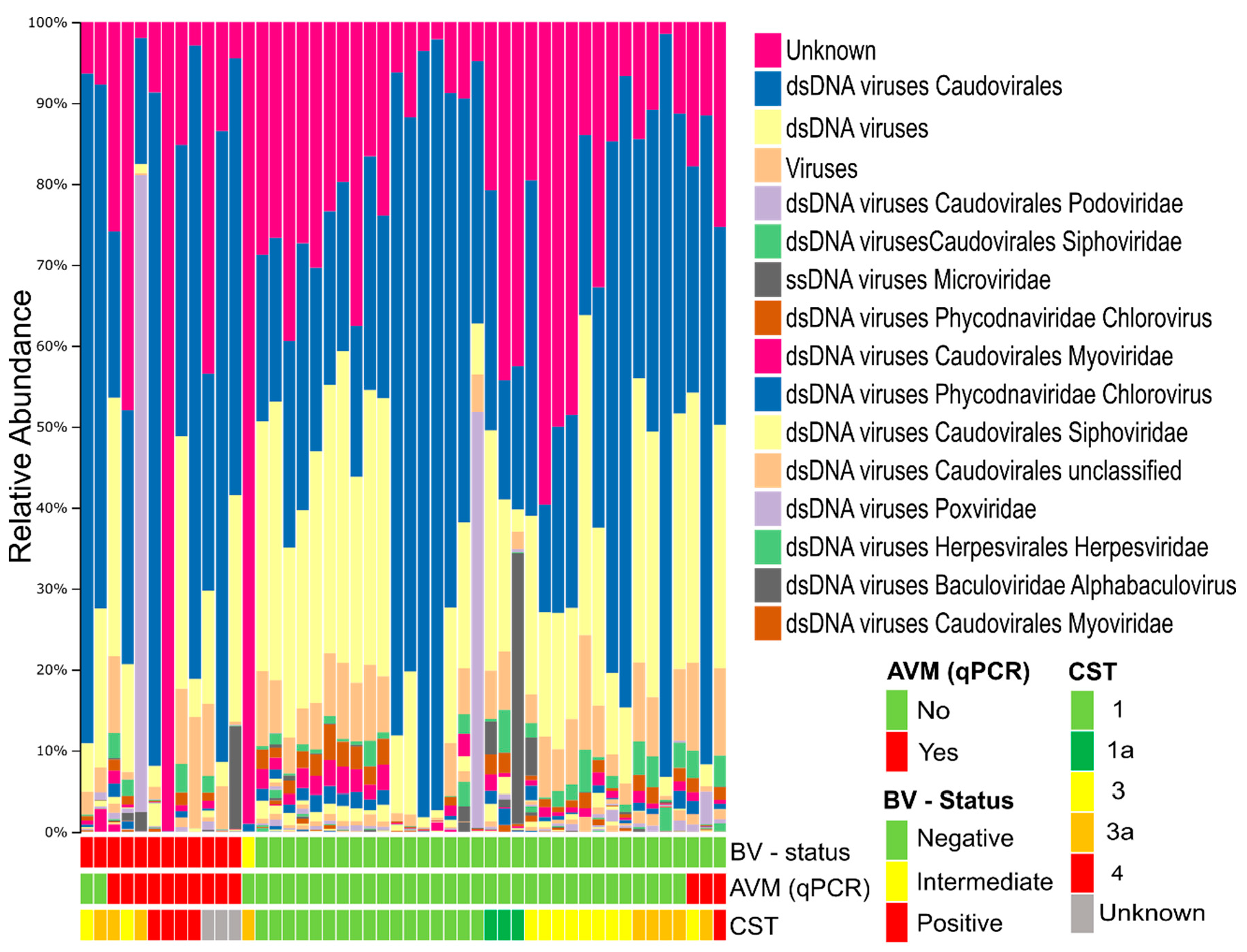
3.2. Composition of the Vaginal Virome
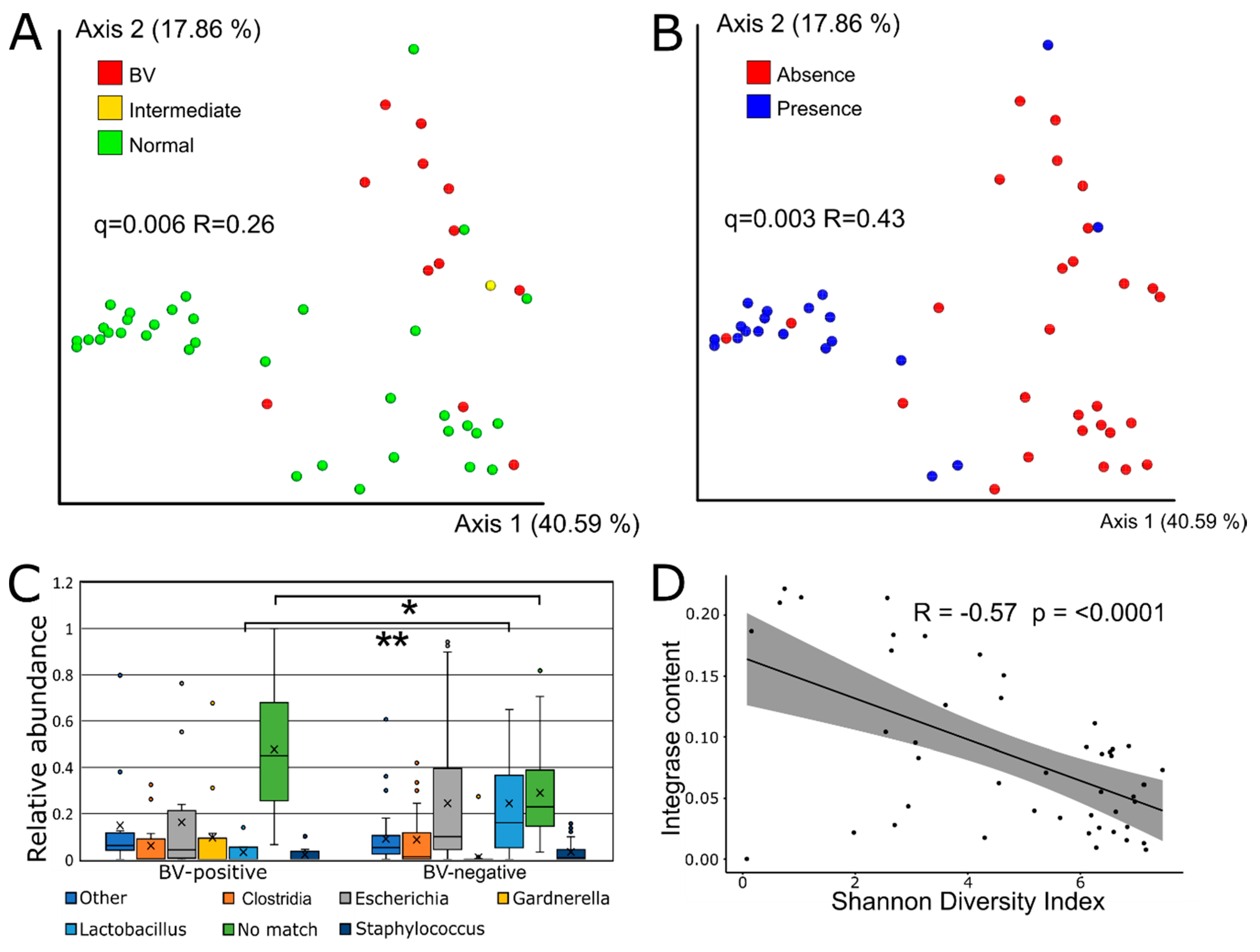
3.3. Viral Beta Diversity is Strongly Correlated with BV-Status
3.4. Sequence-Based Host Prediction and Integrase Content
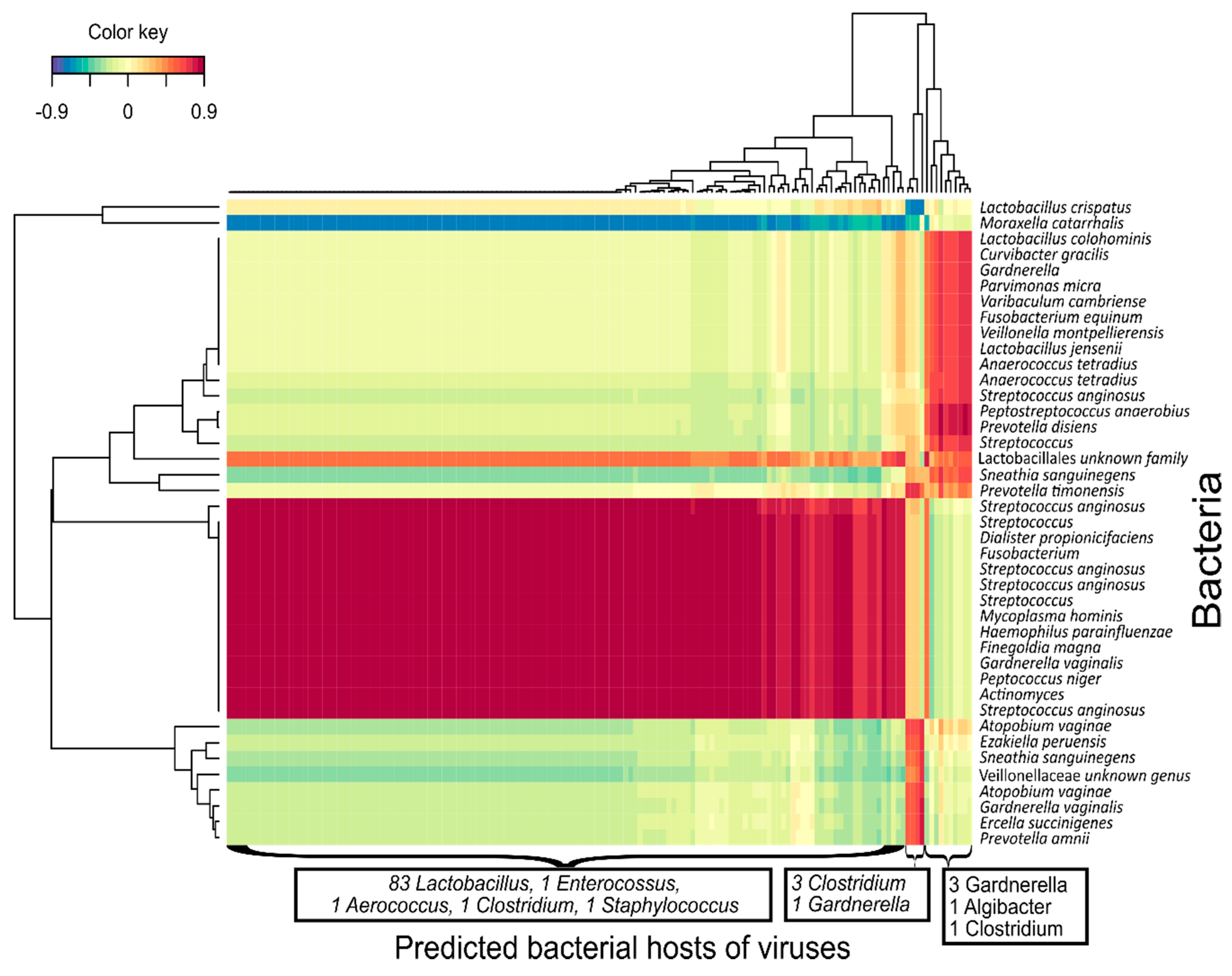
3.5. Bacteriophage-bacteria Interactions
4. Discussion
4.1. Main Findings
4.2. Biological Role of the Virome and Phages in Vaginal Health and Disease
4.3. Comparisons to Other Studies
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Clinical Trials Registration
References
- Nunn, K.L.; Forney, L.J. Unraveling the dynamics of the human vaginal microbiome. Yale J. Biol. Med. 2016, 89, 331–337. [Google Scholar] [PubMed]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal Microbiome: Rethinking Health and Disease Microbiota: Microbial community composition and structure. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.H.; Marrazzo, J.M. The Vaginal Microbiome: Current Understanding and Future Directions. J. Infect. Dis. 2016, 214, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Brown, C.J.; Abdo, Z.; Davis, C.C.; Hansmann, M.A.; Joyce, P.; Foster, J.A.; Forney, L.J. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007, 1, 121–133. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Paul Brooks, J.; Serrano, M.G.; Sheth, N.U.; Girerd, P.H.; Edwards, D.J.; Strauss, J.F.; Jefferson, K.K.; Buck, G.A. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiol. U.K. 2014, 160, 2272–2282. [Google Scholar] [CrossRef]
- Allsworth, J.E.; Peipert, J.F. Prevalence of bacterial vaginosis: 2001-2004 National Health and Nutrition Examination Survey data. Obstet. Gynecol. 2007, 109, 114–120. [Google Scholar] [CrossRef]
- Van de Wijgert, J.H.H.M.; Jespers, V. The global health impact of vaginal dysbiosis. Res. Microbiol. 2017, 168, 859–864. [Google Scholar] [CrossRef]
- Borgdorff, H.; Tsivtsivadze, E.; Verhelst, R.; Marzorati, M.; Jurriaans, S.; Ndayisaba, G.F.; Schuren, F.H.; van de Wijgert, J.H.H.M. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in african women. ISME J. 2014, 8, 1781–1793. [Google Scholar] [CrossRef]
- Haahr, T.; Zacho, J.; Bräuner, M.; Shathmigha, K.; Skov Jensen, J.; Humaidan, P. Reproductive outcome of patients undergoing in vitro fertilization treatment and diagnosed with bacterial vaginosis or abnormal vaginal microbiota: A systematic PRISMA review and meta-analysis. BJOG Int. J. Obstet. Gynaecol. 2018, 126, 200–207. [Google Scholar] [CrossRef]
- Kenyon, C.R.; Buyze, J.; Klebanoff, M.; Brotman, R.M. Association between bacterial vaginosis and partner concurrency: A longitudinal study. Sex. Transm. Infect. 2018, 94, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Marrazzo, J.M.; Fiedler, T.L.; Srinivasan, S.; Mayer, B.T.; Schiffer, J.T.; Fredricks, D.N. Rapid and Profound Shifts in the Vaginal Microbiota Following Antibiotic Treatment for Bacterial Vaginosis. J. Infect. Dis. 2015, 212, 793–802. [Google Scholar] [CrossRef]
- Gray, R.H.; Kigozi, G.; Serwadda, D.; Makumbi, F.; Nalugoda, F.; Watya, S.; Moulton, L.; Chen, M.Z.; Sewankambo, N.K.; Kiwanuka, N.; et al. The effects of male circumcision on female partners’ genital tract symptoms and vaginal infections in a randomized trial in Rakai, Uganda. Am. J. Obstet. Gynecol. 2009, 200, 42.e1–42.e7. [Google Scholar] [CrossRef] [PubMed]
- Schwebke, J.R.; Muzny, C.A.; Josey, W.E. Role of Gardnerella vaginalis in the pathogenesis of bacterial vaginosis: A conceptual model. J. Infect. Dis. 2014, 210, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Colombet, J.; Marteau, P.; Sime-Ngando, T.; Doré, J.; Leclerc, M. Dysbiosis in inflammatory bowel disease: A role for bacteriophages? Gut 2008, 57, 424–425. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Lu, X.-J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179, gutjnl-2018-318131. [Google Scholar] [CrossRef]
- Barksdale, L.; Arden, S.B. Persisting Bacteriophage Infections, Lysogeny, and Phage Conversions. Annu. Rev. Microbiol. 1974, 28, 265–300. [Google Scholar] [CrossRef]
- De Paepe, M.; Leclerc, M.; Tinsley, C.R.; Petit, M.-A. Bacteriophages: An underestimated role in human and animal health? Front. Cell. Infect. Microbiol. 2014, 4, 39. [Google Scholar] [CrossRef]
- Pavlova, S.I.; Tao, L. Induction of vaginal Lactobacillus phages by the cigarette smoke chemical benzo[a]pyrene diol epoxide. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2000, 466, 57–62. [Google Scholar] [CrossRef]
- Jamindar, S.; Polson, S.W.; Furman, M.; Nasko, D.J.; Srinivasiah, S.; Chen, J.; Bhavsar, J.; Dumas, M.; Wommack, K.E. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Ponsero, A.; Thornton, J.; U’Ren, J.M. Phage hunters: Computational strategies for finding phages in large-scale ‘omics datasets. Virus Res. 2018, 244, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and their genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Gosmann, C.; Anahtar, M.N.; Handley, S.A.; Farcasanu, M.; Abu-Ali, G.; Bowman, B.A.; Padavattan, N.; Desai, C.; Droit, L.; Moodley, A.; et al. Lactobacillus-Deficient Cervicovaginal Bacterial Communities Are Associated with Increased HIV Acquisition in Young South African Women. Immunity 2017, 46, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, F.; Wei, W.; Wang, Z.; Dai, J.; Hao, L.; Song, L.; Zhang, X.; Zeng, L.; Du, H.; et al. The metagenome of the female upper reproductive tract. Gigascience 2018, 7. [Google Scholar] [CrossRef]
- Wylie, K.M.; Wylie, T.N.; Cahill, A.G.; Macones, G.A.; Tuuli, M.G.; Stout, M.J. The vaginal eukaryotic DNA virome and preterm birth. Am. J. Obstet. Gynecol. 2018, 219, 189.e1–189.e12. [Google Scholar] [CrossRef]
- Haahr, T.; Humaidan, P.; Elbaek, H.O.; Alsbjerg, B.; Laursen, R.J.; Rygaard, K.; Johannesen, T.B.; Andersen, P.S.; Ng, K.L.; Jensen, J.S. Vaginal microbiota and IVF outcomes: Development of a simple diagnostic tool to predict patients at risk of a poor reproductive outcome. J. Infect. Dis. 2018, 219, 1809–1817. [Google Scholar] [CrossRef]
- Haahr, T.; Ersbøll, A.S.; Karlsen, M.A.; Svare, J.; Sneider, K.; Hee, L.; Weile, L.K.; Ziobrowska-Bech, A.; Østergaard, C.; Jensen, J.S.; et al. Treatment of bacterial vaginosis in pregnancy in order to reduce the risk of spontaneous preterm delivery—A clinical recommendation. Acta Obstet. Gynecol. Scand. 2016, 95, 850–860. [Google Scholar] [CrossRef]
- Robert, P.N.; Marijane, A.K.; Sharon, L.H. Reliability of Diagnosing Bacterial Vaginosis Is Improved by Standardized Method of Gram Stain Interpretation. J. Clin. Microbiol. 1991, 29, 297–301. [Google Scholar] [CrossRef]
- Haahr, T.; Jensen, J.S.; Thomsen, L.; Duus, L.; Rygaard, K.; Humaidan, P. Abnormal vaginal microbiota may be associated with poor reproductive outcomes: A prospective study in IVF patients. Hum. Reprod. 2016, 31, 795–803. [Google Scholar] [CrossRef]
- Conceição-Neto, N.; Zeller, M.; Lefrère, H.; de Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef] [PubMed]
- Marine, R.; McCarren, C.; Vorrasane, V.; Nasko, D.; Crowgey, E.; Polson, S.W.; Wommack, K.E. Caught in the middle with multiple displacement amplification: The myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Ramírez-Guzmán, A.; Taran, Y.; Armienta, M.A. Geochemistry and origin of high-pH thermal springs in the Pacific coast of Guerrero, Mexico. Geofis. Int. 2004, 43, 415–425. [Google Scholar] [CrossRef]
- Galiez, C.; Siebert, M.; Enault, F.; Vincent, J.; Söding, J. WIsH: Who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics 2017, 33, 3113–3114. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Serwer, P. Evolution and the complexity of bacteriophages. Virol. J. 2007, 4, 30. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Zozaya-Hinchliffe, M.; Lillis, R.; Martin, D.H.; Ferris, M.J. Quantitative PCR assessments of bacterial species in women with and without bacterial vaginosis. J. Clin. Microbiol. 2010, 48, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Dynamic viral populations in hypersaline systems as revealed by metagenomic assembly. Appl. Environ. Microbiol. 2012, 78, 6309–6320. [Google Scholar] [CrossRef] [PubMed]
- Squier, A.H.; Hodgson, D.A.; Keely, B.J. Sedimentary pigments as markers for environmental change in an Antarctic lake. Org. Geochem. 2002, 33, 1655–1665. [Google Scholar] [CrossRef]
- Maslov, S.; Sneppen, K. Well-temperate phage: Optimal bet-hedging against local environmental collapses. Sci. Rep. 2015, 5, 10523. [Google Scholar] [CrossRef] [PubMed]
- Muzny, C.A.; Lensing, S.Y.; Aaron, K.J.; Schwebke, J.R. Incubation period and risk factors support sexual transmission of bacterial vaginosis in women who have sex with women. Sex. Transm. Infect. 2019, 95, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Bae, J.W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 2018, 12, 1127–1141. [Google Scholar] [CrossRef]
- Vostrov, A.A.; Vostrukhina, O.A.; Svarchevsky, A.N.; Rybchin, V.N. Proteins responsible for lysogenic conversion caused by coliphages N15 and phi80 are highly homologous. J. Bacteriol. 1996, 178, 1484–1486. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human virome and disease: High-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef]
- Dezzutti, C.S.; Hendrix, C.W.; Marrazzo, J.M.; Pan, Z.; Wang, L.; Louissaint, N.; Kalyoussef, S.; Torres, N.M.; Hladik, F.; Parikh, U.; et al. Performance of Swabs, Lavage, and Diluents to Quantify Biomarkers of Female Genital Tract Soluble Mucosal Mediators. PLoS ONE 2011, 6, e23136. [Google Scholar] [CrossRef]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.; Baldridge, M.T.; Handley, S.A. Phages and human health: More than idle hitchhikers. Viruses 2019, 11, 587. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patients, No. (%) a (n = 48) | |
|---|---|
| Age, median (range), y | 29 (23–41) |
| Body mass index, median(range) b | 31 (17.5–41) |
| Ethnicity | |
| Caucasian | 83 (40) |
| Eastern European | 8 (4) |
| Other | 6 (3) |
| Asian | 2 (1) |
| Cause of infertility | |
| Male factor | 58 (28) |
| Single | 13 (6) |
| Lesbian | 3 (2) |
| Nugent Group c | |
| BV | 25 (12) |
| Normal | 73 (35) |
| Intermediate | 2 (1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakobsen, R.R.; Haahr, T.; Humaidan, P.; Jensen, J.S.; Kot, W.P.; Castro-Mejia, J.L.; Deng, L.; Leser, T.D.; Nielsen, D.S. Characterization of the Vaginal DNA Virome in Health and Dysbiosis. Viruses 2020, 12, 1143. https://doi.org/10.3390/v12101143
Jakobsen RR, Haahr T, Humaidan P, Jensen JS, Kot WP, Castro-Mejia JL, Deng L, Leser TD, Nielsen DS. Characterization of the Vaginal DNA Virome in Health and Dysbiosis. Viruses. 2020; 12(10):1143. https://doi.org/10.3390/v12101143
Chicago/Turabian StyleJakobsen, Rasmus Riemer, Thor Haahr, Peter Humaidan, Jørgen Skov Jensen, Witold Piotr Kot, Josue Leonardo Castro-Mejia, Ling Deng, Thomas Dyrmann Leser, and Dennis Sandris Nielsen. 2020. "Characterization of the Vaginal DNA Virome in Health and Dysbiosis" Viruses 12, no. 10: 1143. https://doi.org/10.3390/v12101143
APA StyleJakobsen, R. R., Haahr, T., Humaidan, P., Jensen, J. S., Kot, W. P., Castro-Mejia, J. L., Deng, L., Leser, T. D., & Nielsen, D. S. (2020). Characterization of the Vaginal DNA Virome in Health and Dysbiosis. Viruses, 12(10), 1143. https://doi.org/10.3390/v12101143