Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response that Blocks Influenza A Virus Replication

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Influenza A Viruses
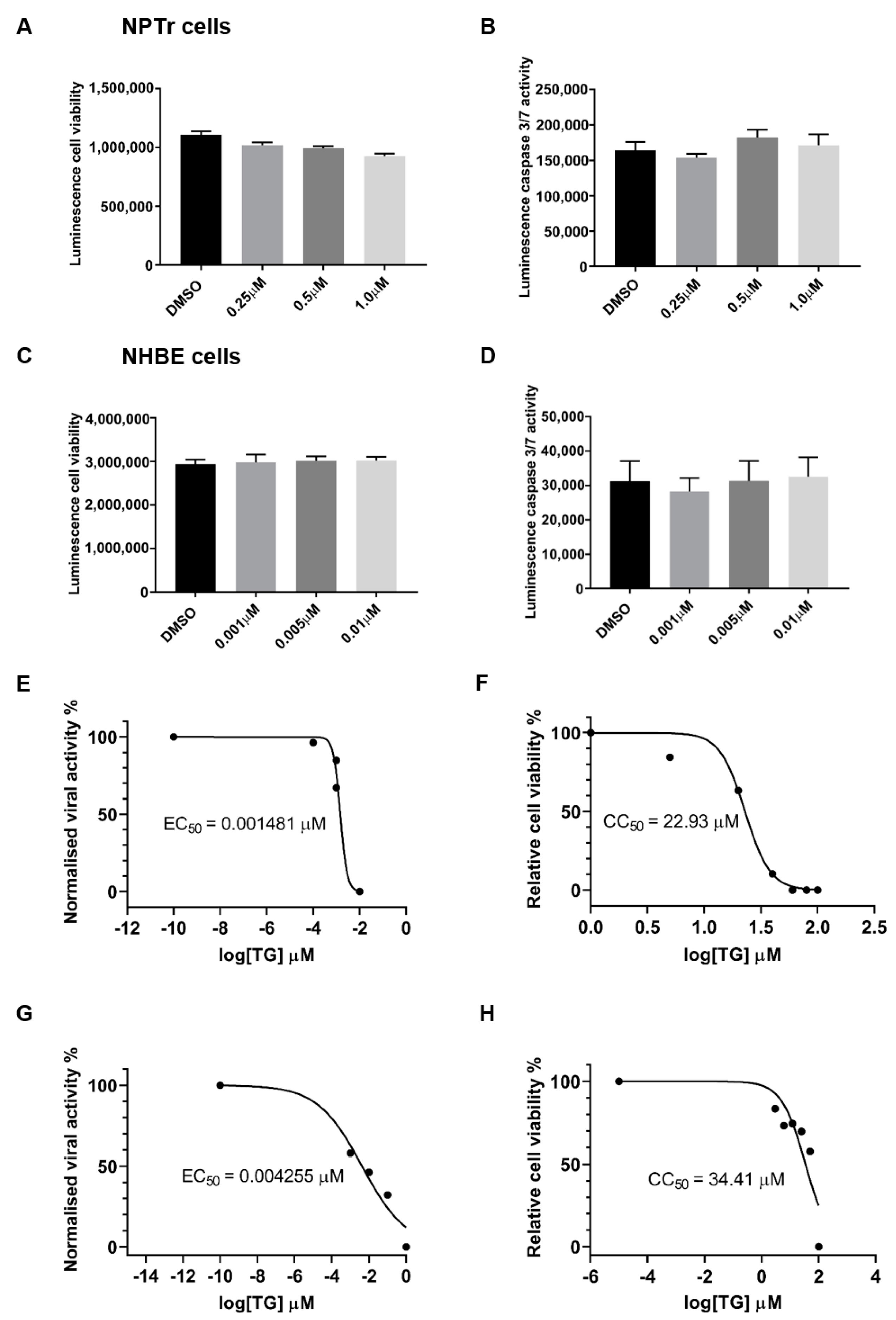
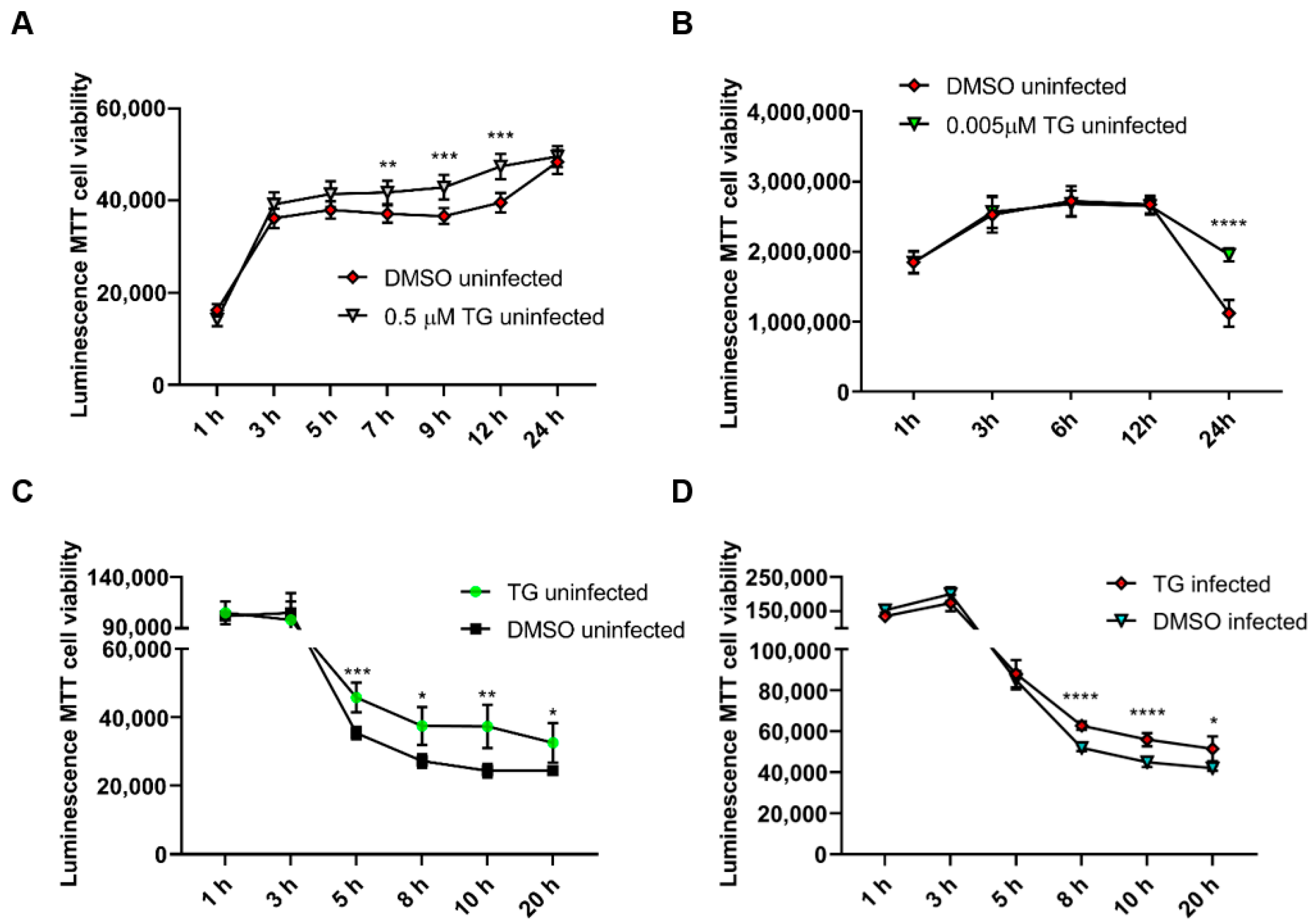
2.2. Cell Viability and Caspase 3/7 Assays
2.3. Chemical Priming of Cells
2.4. Infection and Progeny Virus Quantification
2.5. RNA Preparation and Real-Time RT-PCR
2.6. Western Blotting
2.7. Confocal microscopy
2.8. Influenza Virus Challenge in Mice
2.9. RNA-Sequencing
2.10. Quantification and Statistical Analysis
2.11. Ethics Statement
3. Results
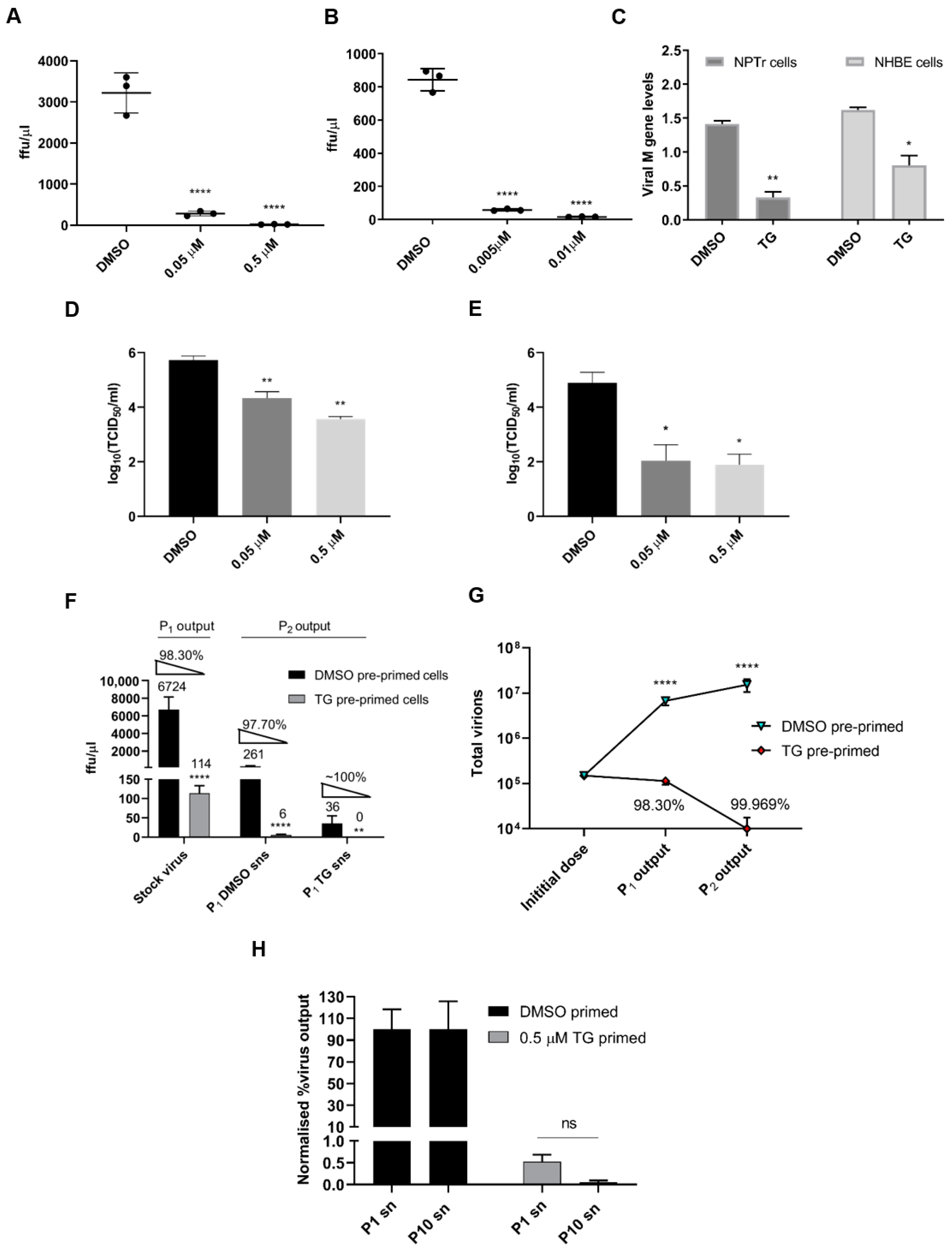
3.1. Non-Cytotoxic Application of TG Blocks Influenza A Virus Replication in Respiratory Epithelial Cells
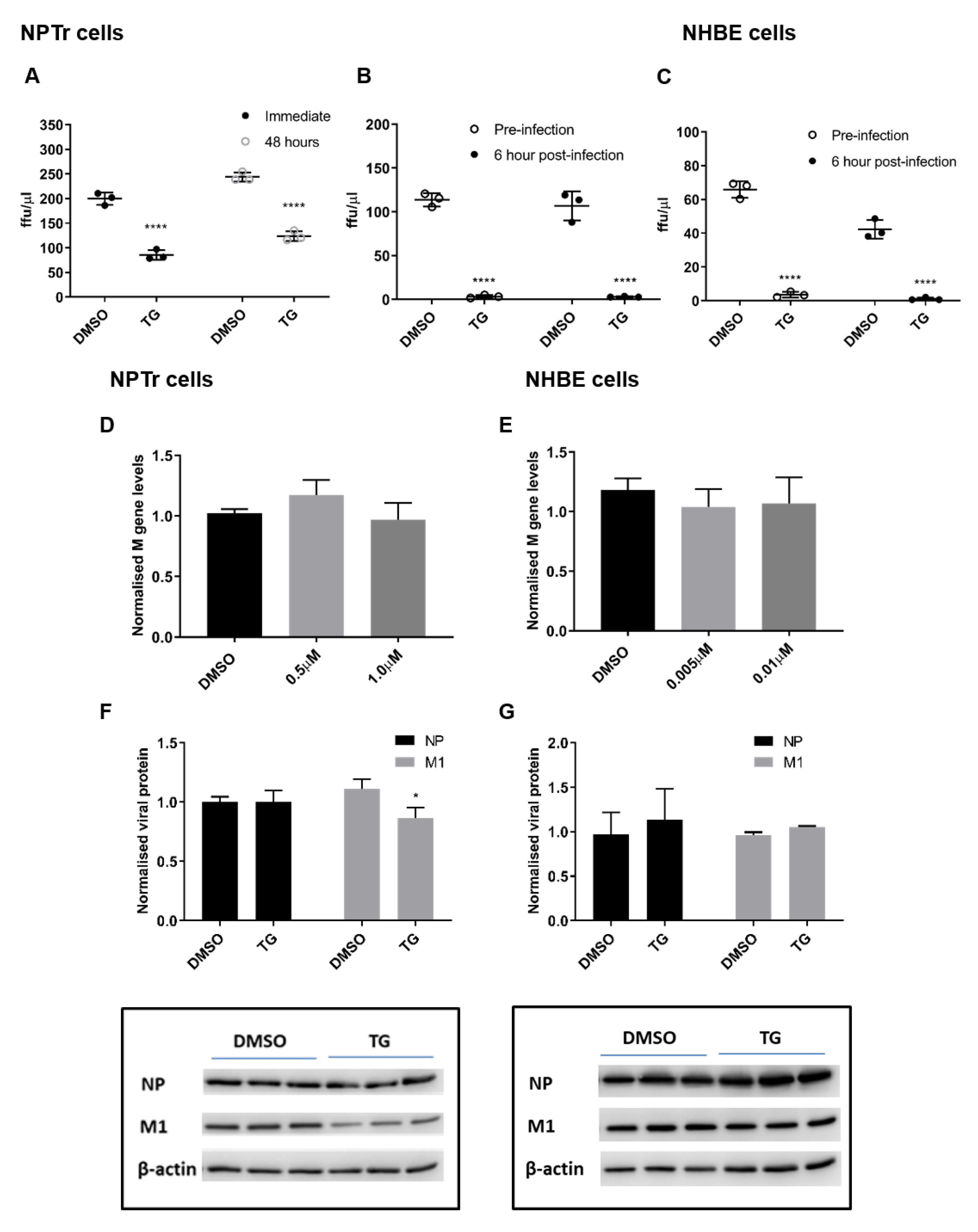
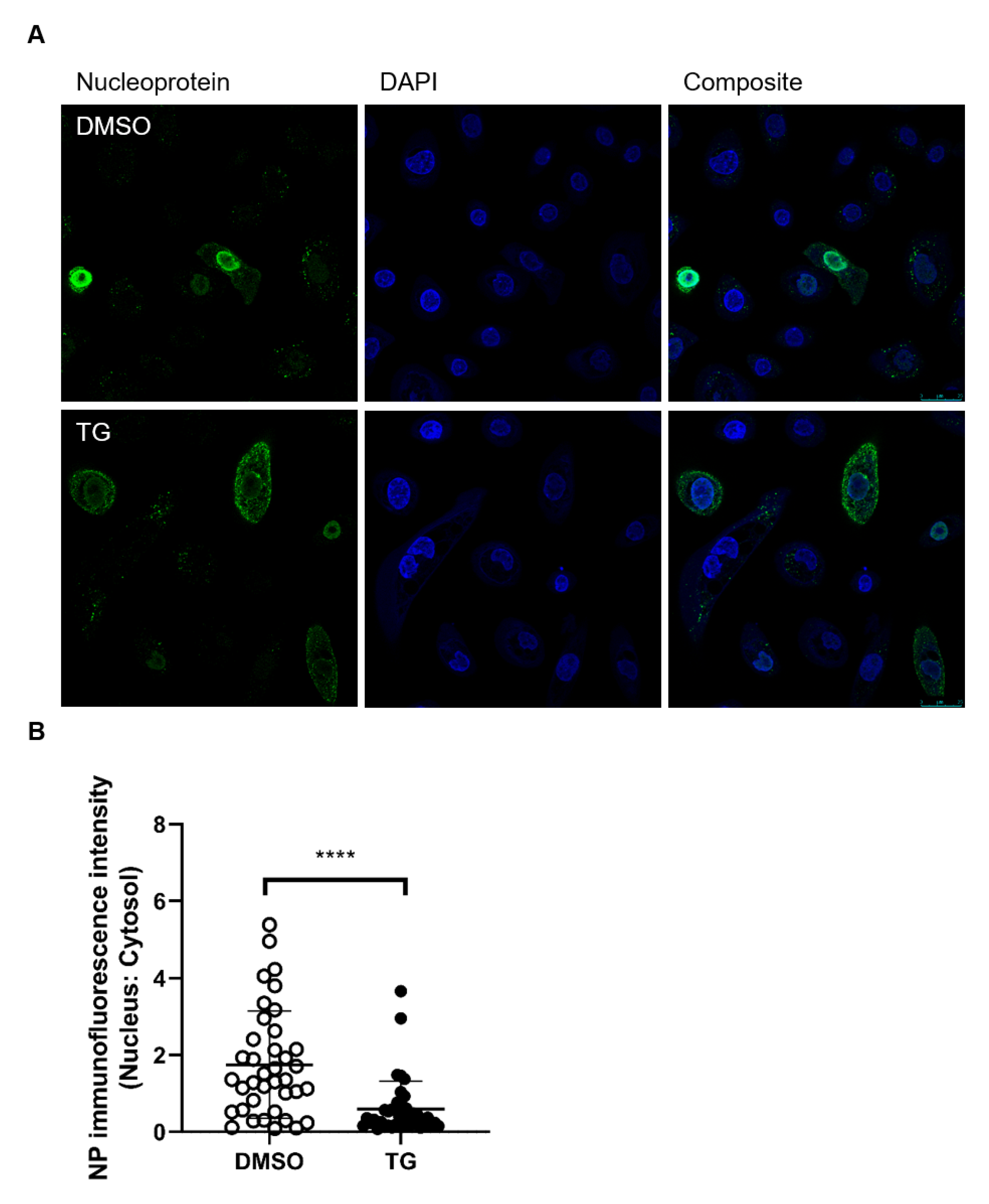
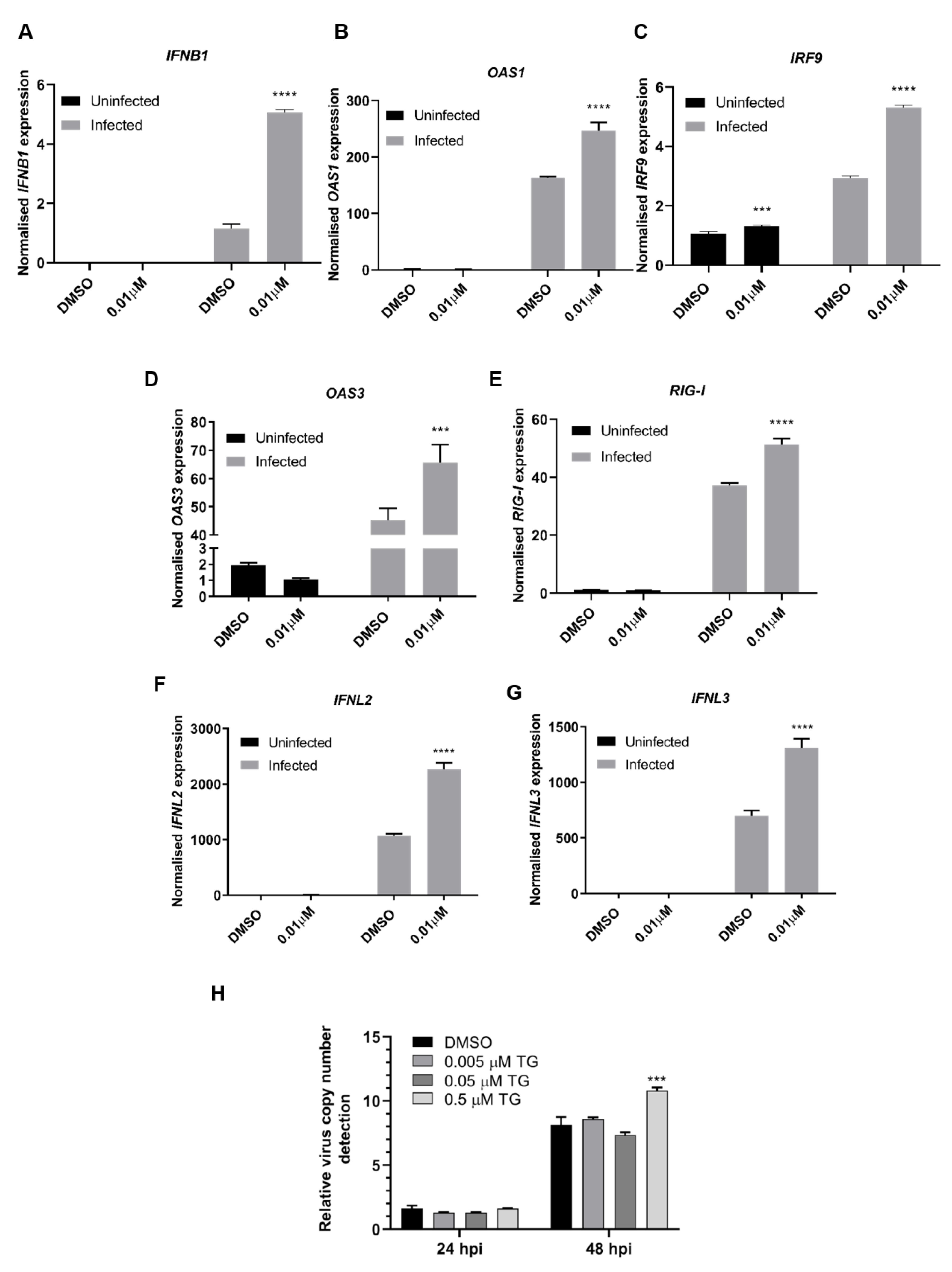
3.2. TG Promptly Induces an Extended Antiviral State Targeting Influenza Virus Post-Translationally
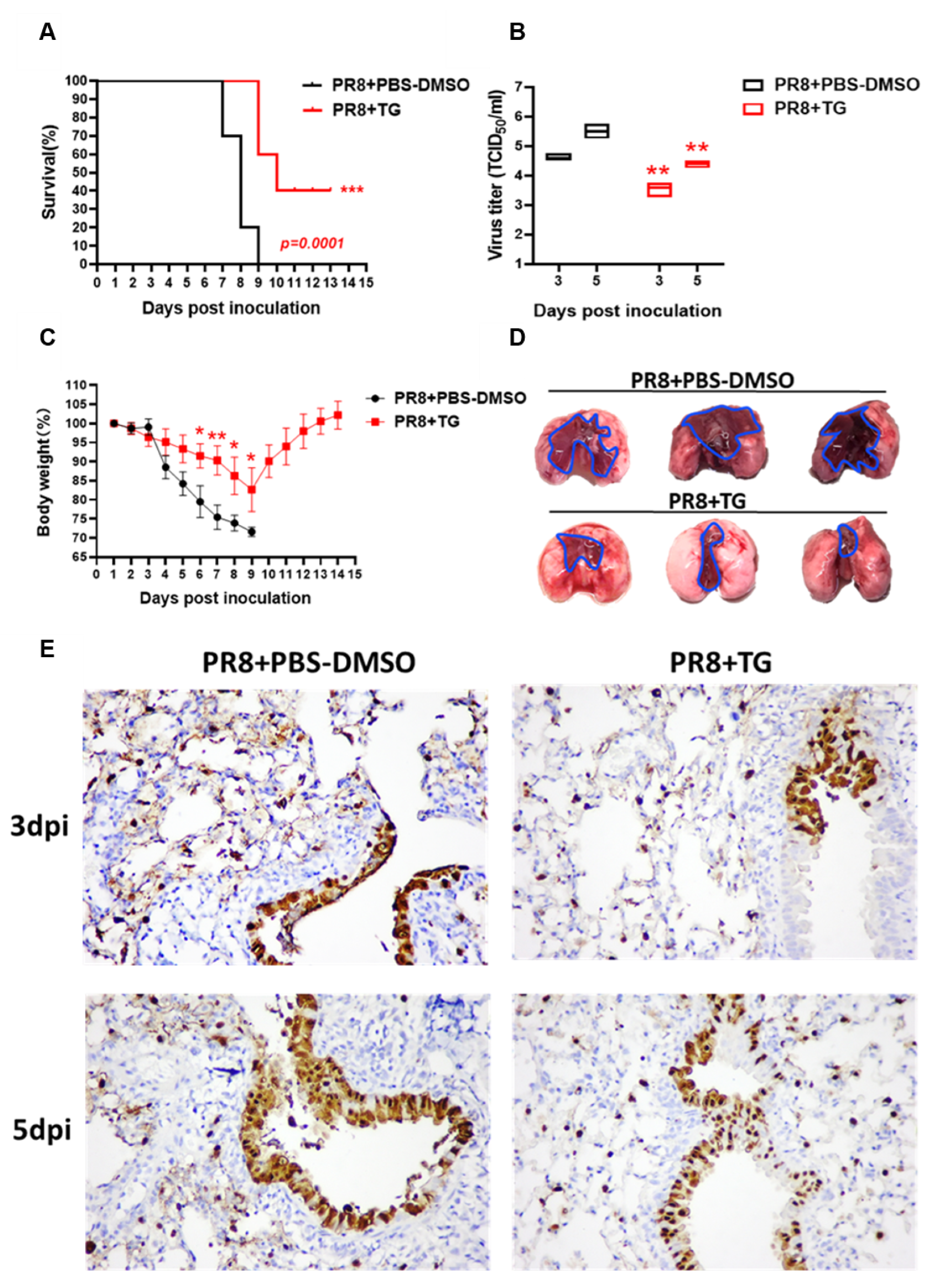
3.3. TG in Mice Confers Protection Against Lethal Virus Challenge
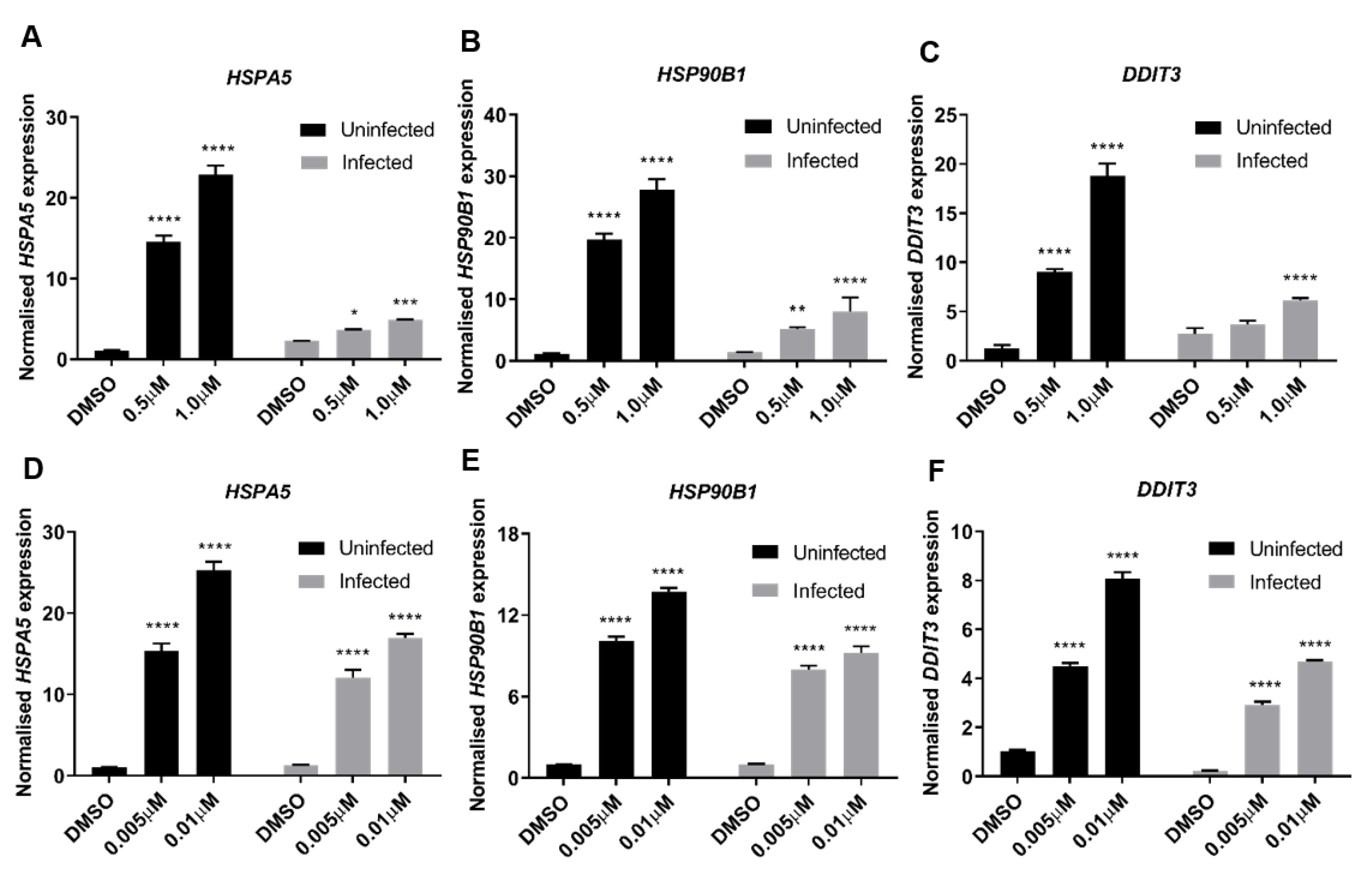
3.4. ER Stress Is a Dominant Driver of Host Antiviral Response to TG Priming
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Goldhill, D.H.; Velthuis, A.J.W.T.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef]
- Takashita, E.; Kawakami, C.; Ogawa, R.; Morita, H.; Fujisaki, S.; Shirakura, M.; Miura, H.; Nakamura, K.; Kishida, N.; Kuwahara, T.; et al. Influenza A(H3N2) virus exhibiting reduced susceptibility to baloxavir due to a polymerase acidic subunit I38T substitution detected from a hospitalised child without prior baloxavir treatment, Japan, January 2019. Eurosurveillance 2019, 24, 1900170. [Google Scholar] [CrossRef]
- Yong, H.Y.; Luo, D. RIG-I-Like Receptors as Novel Targets for Pan-Antivirals and Vaccine Adjuvants Against Emerging and Re-Emerging Viral Infections. Front. Immunol. 2018, 9, 1379. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Hou, W.; Dakshanamurthy, S.; Tang, Q. Host targeted antiviral (HTA): Functional inhibitor compounds of scaffold protein RACK1 inhibit herpes simplex virus proliferation. Oncotarget 2019, 10, 3209–3226. [Google Scholar] [CrossRef] [PubMed]
- Warfield, K.L.; Schaaf, K.R.; Dewald, L.E.; Spurgers, K.B.; Wang, W.; Stavale, E.; Mendenhall, M.; Shilts, M.H.; Stockwell, T.B.; Barnard, D.L.; et al. Lack of selective resistance of influenza A virus in presence of host-targeted antiviral, UV-4B. Sci. Rep. 2019, 9, 7484. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2017, 17, 35–56. [Google Scholar] [CrossRef]
- Meyniel-Schicklin, L.; De Chassey, B.; André, P.; Lotteau, V. Viruses and interactomes in translation. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar]
- Krebs, J.; Agellon, L.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.L.; Monick, M.M. Influenza A Viral Replication Is Blocked by Inhibition of the Inositol-requiring Enzyme 1 (IRE1) Stress Pathway. J. Biol. Chem. 2011, 287, 4679–4689. [Google Scholar] [CrossRef] [PubMed]
- Ranjitha, H.B.; Ammanathan, V.; Guleria, N.; Hosamani, M.; Sreenivasa, B.P.; Dhanesh, V.V.; Santhoshkumar, R.; Sagar, B.K.C.; Mishra, B.P.; Singh, R.K.; et al. Foot-and-mouth disease virus induces PERK mediated autophagy to suppress antiviral interferon response. J. Cell Sci. 2020, 134, 240622. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Zeng, L.; Tian, A.; Bomkamp, A.; Rivera, D.; Gutman, D.; Barber, G.N.; Olson, J.K.; Smith, J.A. Endoplasmic Reticulum Stress Regulates the Innate Immunity Critical Transcription Factor IRF3. J. Immunol. 2012, 189, 4630–4639. [Google Scholar] [CrossRef] [PubMed]
- So, J.-S. Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 2018, 41, 705–716. [Google Scholar]
- Han, S.; Mao, L.; Liao, Y.; Sun, S.; Zhang, Z.; Mo, Y.; Liu, H.; Zhi, X.; Lin, S.; Seo, H.S.; et al. Sec62 Suppresses Foot-and-Mouth Disease Virus Proliferation by Promotion of IRE1α-RIG-I Antiviral Signaling. J. Immunol. 2019, 203, 429–440. [Google Scholar] [CrossRef]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef]
- Kuchipudi, S.V.; Dunham, S.P.; Nelli, R.; White, G.A.; Coward, V.J.; Slomka, M.J.; Brown, I.H.; Chang, K.-C. Rapid death of duck cells infected with influenza: A potential mechanism for host resistance to H5N1. Immunol. Cell Biol. 2011, 90, 116–123. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Lupfer, C.; Thomas, P.G.; Anand, P.K.; Vogel, P.; Milasta, S.; Martinez, J.; Huang, G.; Green, M.; Kundu, M.; Chi, H.; et al. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013, 14, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Ferrari, M.; Scalvini, A.; Losio, M.N.; Corradi, A.; Soncini, M.; Bignotti, E.; Milanesi, E.; Ajmone-Marsan, P.; Barlati, S.; Bellotti, D.; et al. Establishment and characterization of two new pig cell lines for use in virological diagnostic laboratories. J. Virol. Methods 2003, 107, 205–212. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef]
- Lencer, W.I.; DeLuca, H.; Grey, M.J.; Cho, J.A. Innate immunity at mucosal surfaces: The IRE1-RIDD-RIG-I pathway. Trends Immunol. 2015, 36, 401–409. [Google Scholar] [CrossRef]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of Interferon Production and of Rubella Virus Interference in a Line of African Green Monkey Kidney Cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Mosca, J.D.; Pitha, P.M. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol. Cell. Biol. 1986, 6, 2279–2283. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Chang, C.; Li, L.; Klenk, C.; Cheng, J.; Chen, Y.; Xia, N.; Shu, Y.; Chen, Z.; Gabriel, G.; et al. Sumoylation of Influenza A Virus Nucleoprotein Is Essential for Intracellular Trafficking and Virus Growth. J. Virol. 2014, 88, 9379–9390. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Jeng, K.-S.; Lai, M.M.-C. The SUMOylation of Matrix Protein M1 Modulates the Assembly and Morphogenesis of Influenza A Virus. J. Virol. 2011, 85, 6618–6628. [Google Scholar] [CrossRef]
- Su, W.-C.; Yu, W.-Y.; Huang, S.-H.; Lai, M.M.C. Ubiquitination of the Cytoplasmic Domain of Influenza A Virus M2 Protein is Crucial for Production of Infectious Virus Particles. J. Virol. 2017, 92, JVI.01972-17. [Google Scholar] [CrossRef]
- Checkmahomed, L.; M’Hamdi, Z.; Carbonneau, J.; Venable, M.-C.; Baz, M.; Abed, Y.; Boivin, G. Impact of the baloxavir-resistant polymerase acid (PA) I38T substitution on the fitness of contemporary influenza A(H1N1)pdm09 and A(H3N2) strains. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Uehara, T.; Hayden, F.G.; Kawaguchi, K.; Omoto, S.; Hurt, A.C.; De Jong, M.D.; Hirotsu, N.; Sugaya, N.; Lee, N.; Baba, K.; et al. Treatment-Emergent Influenza Variant Viruses with Reduced Baloxavir Susceptibility: Impact on Clinical and Virologic Outcomes in Uncomplicated Influenza. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Presloid, J.B.; Novella, I.S. RNA Viruses and RNAi: Quasispecies Implications for Viral Escape. Viruses 2015, 7, 3226–3240. [Google Scholar] [CrossRef]
- Heaton, S.M. Harnessing host–virus evolution in antiviral therapy and immunotherapy. Clin. Transl. Immunol. 2019, 8, e1067. [Google Scholar] [CrossRef]
- Aljofan, M.; Lo, M.K.; Rota, P.A.; Michalski, W.P.; Mungall, B.A. Off Label Antiviral Therapeutics for Henipaviruses: New Light Through Old Windows. J. Antivir. Antiretrovir. 2010, 2, 1–10. [Google Scholar]
- Landeras-Bueno, S.; Fernàndez, Y.; Falcon, A.; Oliveros, J.C.; Ortín, J. Chemical Genomics Identifies the PERK-Mediated Unfolded Protein Stress Response as a Cellular Target for Influenza Virus Inhibition. mBio 2016, 7, 00085-16. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.; Sesaki, H.; Kang, S.-J. Intracellular calcium is a rheostat for the STING signaling pathway. Biochem. Biophys. Res. Commun. 2018, 500, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Klenk, C.; Liu, B.; Keiner, B.; Cheng, J.; Zheng, B.J.; Li, L.; Han, Q.; Wang, C.; Li, T.; et al. Modification of non-structural protein 1 of influenza A virus by SUMO. J. Virol. 2011, 85, 1086–1098. [Google Scholar] [CrossRef] [PubMed]
- Kirui, J.; Mondal, A.; Mehle, A. Ubiquitination up-regulates influenza virus polymerase function. J. Virol. 2016, 90, 10906–10914. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense Primer (5′–3′) | Antisense Primer (5′–3′) |
|---|---|---|
| 18S ribosomal RNA (universal) | ACGGCTACCACATCCAAGGA | CCAATTACAGGGCCTCGAAA |
| RIG-I (human) | GAAGGCATTGACATTGCACAGT | TGGTTTGGATCATTTTGATGACA |
| IFNL2 (human) | GACCCAGCCCTGGTGGAC | GCTGGATACAGGCCCGGAA |
| IFNL3 (human) | GACCCAGCCCTGGGGGAT | GCTGGATACAGGCCCGGAG |
| RIG-I (pig) | CCCTGGTTTAGGGACGATGAG | AACAGGAACTGGAGAAAAGTGA |
| OAS1 (pig) | GAGCTGCAGCGAGACTTCCT | GGCGGATGAGGCTCTTCA |
| M-gene (USSR H1N1) | AGATGAGCCTTCTAACCGAGGTCG | TGCAAAAACATCTTCAAGTCTCTG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goulding, L.V.; Yang, J.; Jiang, Z.; Zhang, H.; Lea, D.; Emes, R.D.; Dottorini, T.; Pu, J.; Liu, J.; Chang, K.-C. Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response that Blocks Influenza A Virus Replication. Viruses 2020, 12, 1093. https://doi.org/10.3390/v12101093
Goulding LV, Yang J, Jiang Z, Zhang H, Lea D, Emes RD, Dottorini T, Pu J, Liu J, Chang K-C. Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response that Blocks Influenza A Virus Replication. Viruses. 2020; 12(10):1093. https://doi.org/10.3390/v12101093
Chicago/Turabian StyleGoulding, Leah V., Jiayun Yang, Zhimin Jiang, Hongyu Zhang, Daniel Lea, Richard D. Emes, Tania Dottorini, Juan Pu, Jinhua Liu, and Kin-Chow Chang. 2020. "Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response that Blocks Influenza A Virus Replication" Viruses 12, no. 10: 1093. https://doi.org/10.3390/v12101093
APA StyleGoulding, L. V., Yang, J., Jiang, Z., Zhang, H., Lea, D., Emes, R. D., Dottorini, T., Pu, J., Liu, J., & Chang, K.-C. (2020). Thapsigargin at Non-Cytotoxic Levels Induces a Potent Host Antiviral Response that Blocks Influenza A Virus Replication. Viruses, 12(10), 1093. https://doi.org/10.3390/v12101093