Network Controllability-Based Prioritization of Candidates for SARS-CoV-2 Drug Repositioning



Abstract
1. Introduction
2. Methods
2.1. Protein–Protein Interaction Network Construction
2.2. Robust Classification
2.3. Global Classification
3. Results
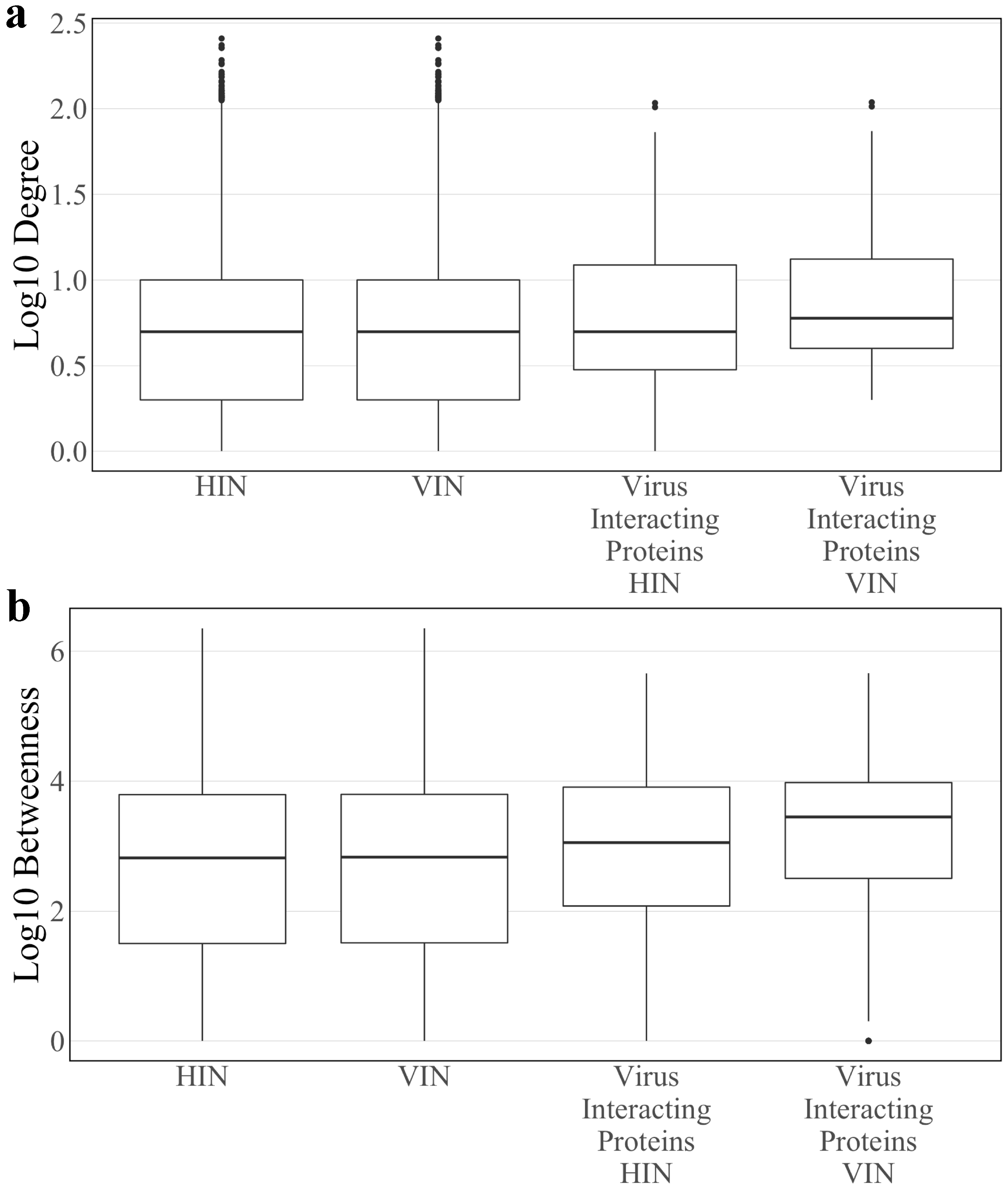
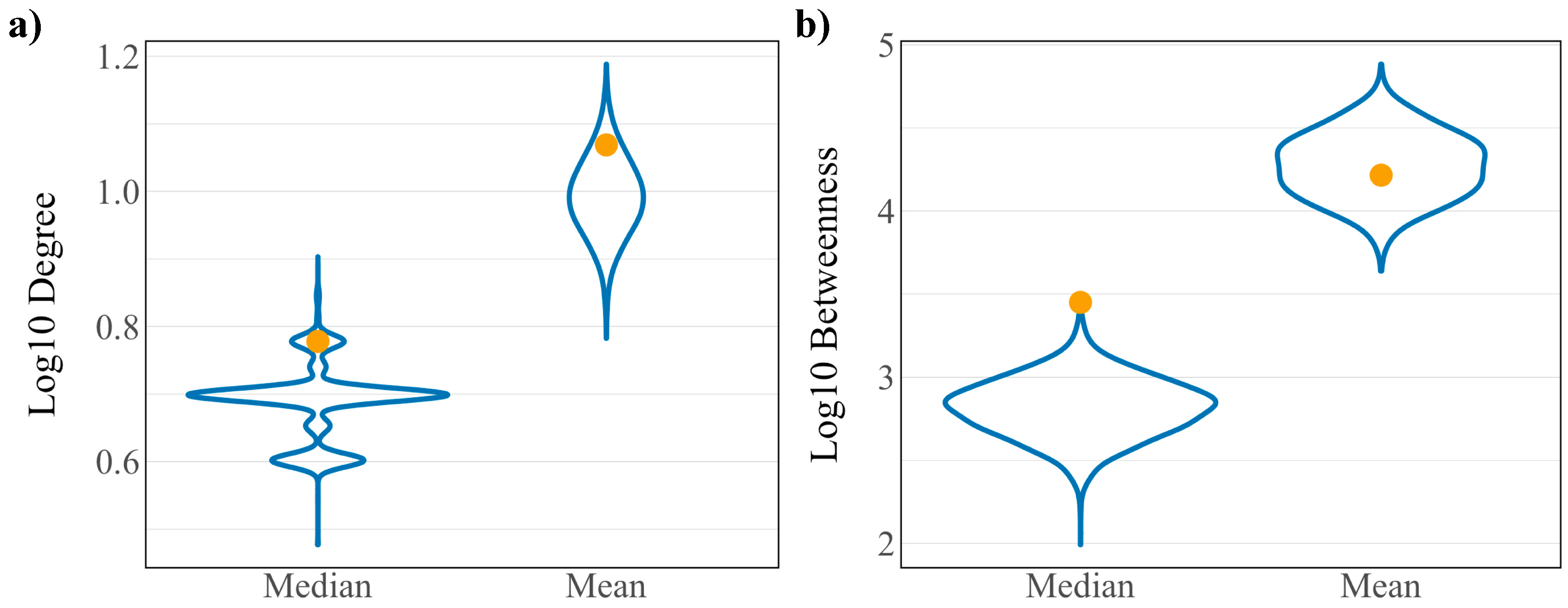
3.1. Topology of the Host Interaction Network and Virus Integrated Network
3.2. Driver Proteins
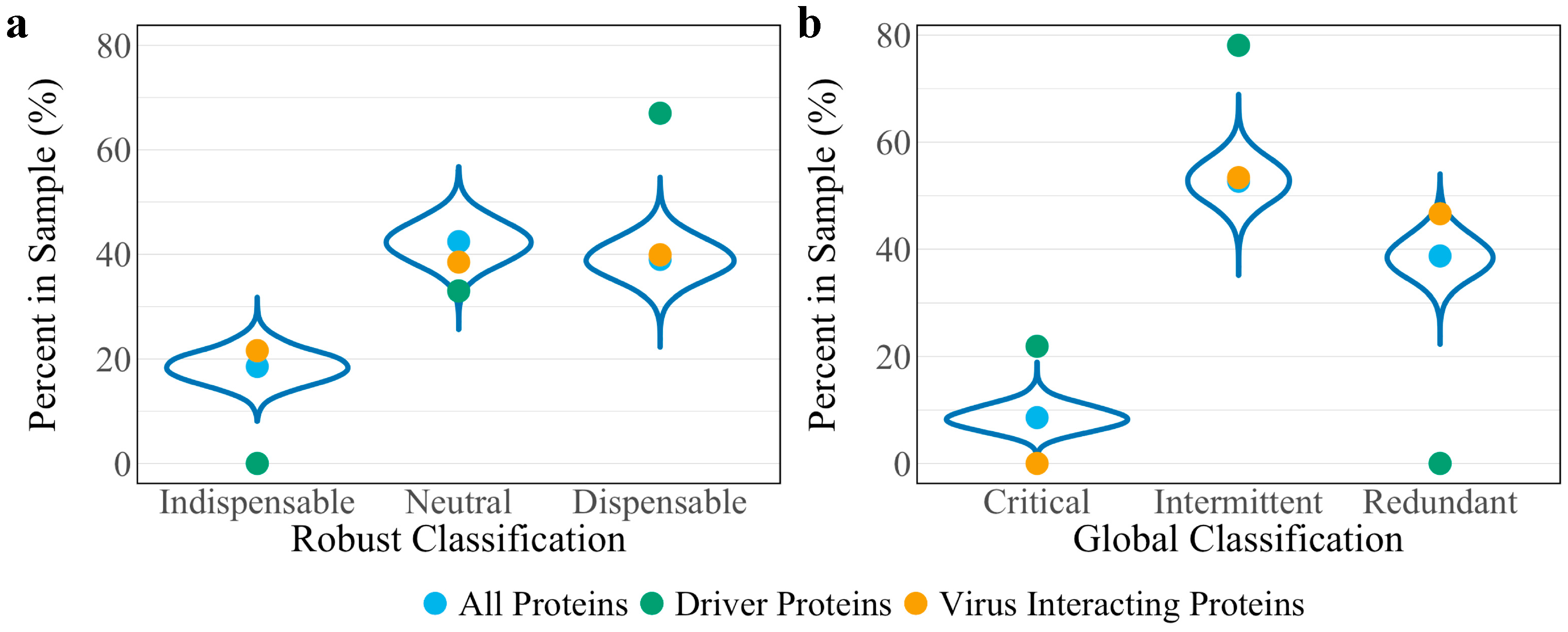
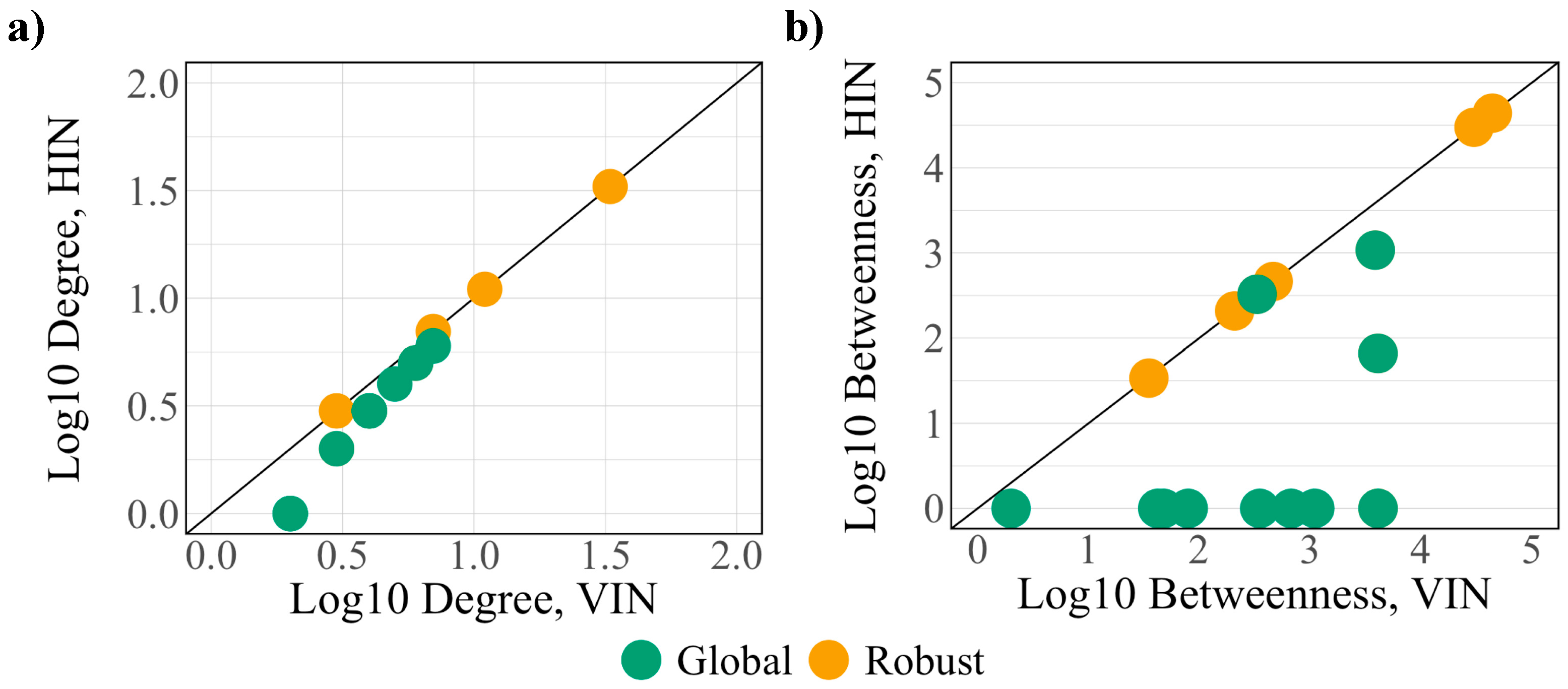
3.3. Robust Controllability
3.4. Global Controllability
3.5. Controllability-Guided Drug Targeting and Repurposing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Coronavirus disease (COVID-19) Weekly Epidemiological Update. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200831-weekly-epi-update-3.pdf?sfvrsn=d7032a2a_4 (accessed on 25 September 2020).
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; Decroly, E.; de Lamballerie, X.; Coutard, B. In vitro screening of a FDA approved chemical library reveals potential inhibitors of SARS-CoV-2 replication. Sci. Rep. 2020, 10, 1–8. [Google Scholar]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Burgstaller, S.; Pache, L.; De Jesus, P.; Hull, M.V.; Chang, M.; et al. A Large-scale Drug Repositioning Survey for SARS-CoV-2 Antivirals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bernhard, E.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; Geisslinger, G.; et al. Identification of inhibitors of SARS-CoV-2 in-vitro cellular toxicity in human (Caco-2) cells using a large scale drug repurposing collection. Res. Sq. 2020. [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. Nature 2020, 583, 1–13. [Google Scholar]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14–18. [Google Scholar] [PubMed]
- Perrin-Cocon, L.; Diaz, O.; Jacquemin, C.; Barthel, V.; Ogire, E.; Ramière, C.; André, P.; Lotteau, V.; Vidalain, P.O. The current landscape of coronavirus-host protein-protein interactions. J. Transl. Med. 2020, 18, 1–15. [Google Scholar]
- Cava, C.; Bertoli, G.; Castiglioni, I. A protein interaction map identifies existing drugs targeting SARS-CoV-2. BMC Pharmacol. Toxicol. 2020, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, E.E.; Alcorn, J.F.; Hase, T.; Shoemaker, J.E. A dual controllability analysis of influenza virus-host protein-protein interaction networks for antiviral drug target discovery. BMC Bioinform. 2019, 20, 297. [Google Scholar] [CrossRef]
- Sontag, E. Mathematical Control Theory: Deterministic Finite Dimensional Systems; Springer Science & Business Media: Berlin, Germany, 1998; ISBN 978-1-4612-0577-7. [Google Scholar]
- Jia, T.; Barabási, A.L. Control capacity and a random sampling method in exploring controllability of complex networks. Sci. Rep. 2013, 3, srep02354. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Slotine, J.J.; Barabási, A.L. Controllability of complex networks. Nature 2011, 473, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Liu, Y.-Y.; Csóka, E.; Pósfai, M.; Slotine, J.-J.; Barabási, A.-L. Emergence of bimodality in controlling complex networks. Nat. Commun. 2013, 4, 2002. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Stelzl, U.; Foulle, R.; Plassmann, S.; Zenkner, M.; Timm, J.; Assmus, H.E.; Andrade-Navarro, M.A.; Wanker, E.E. A directed protein interaction network for investigating intracellular signal transduction. Sci. Signal. 2011, 4, rs8. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Finley, R.L. Combining multiple positive training sets to generate confidence scores for protein-protein interactions. Bioinformatics 2008, 25, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Hopcroft, J.E.; Karp, R.M. An $n^{5/2} $ Algorithm for Maximum Matchings in Bipartite Graphs. SIAM J. Comput. 1973, 2, 225–231. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, T.; Pu, Y. Input graph: The hidden geometry in controlling complex networks. Sci. Rep. 2016, 6, 38209. [Google Scholar] [CrossRef]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM Protein-Mediated Regulation of Inflammatory and Innate Immune Signaling and Its Association with Antiretroviral Activity. J. Virol. 2012, 87, 257–272. [Google Scholar] [CrossRef]
- Jiang, M.X.; Hong, X.; Liao, B.B.; Shi, S.Z.; Lai, X.F.; Zheng, H.Y.; Xie, L.; Wang, Y.; Wang, X.L.; Xin, H.B.; et al. Expression profiling of TRIM protein family in THP1-derived macrophages following TLR stimulation. Sci. Rep. 2017, 7, 42781. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 2015, 6. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress- inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Hsiung, B.; Pestal, K.; Procyk, E.; Raulet, D.H. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which controlcell cycle entry. J. Exp. Med. 2012, 209, 2409–2422. [Google Scholar] [CrossRef]
- Samarajiwa, S.A.; Forster, S.; Auchettl, K.; Hertzog, P.J. INTERFEROME: The database of interferon regulated genes. Nucleic Acids Res. 2009, 37, D852–D857. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A.; Rosen, H. Cytokine storm plays a direct role in the morbidity and mortality from influenza virus infection and is chemically treatable with a single sphingosine-1-phosphate agonist molecule. Curr. Top. Microbiol. Immunol. 2014, 378, 129–147. [Google Scholar]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020, 1–6. [Google Scholar] [CrossRef]
- Scarselli, E.; Ansuini, H.; Cerino, R.; Roccasecca, R.M.; Acali, S.; Filocamo, G.; Traboni, C.; Nicosia, A.; Cortese, R.; Vitelli, A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002, 21, 5017–5025. [Google Scholar] [CrossRef]
- van Nieuwenhuijze, A.; Burton, O.; Lemaitre, P.; Denton, A.E.; Cascalho, A.; Goodchild, R.E.; Malengier-Devlies, B.; Cauwe, B.; Linterman, M.A.; Humblet-Baron, S.; et al. Mice Deficient in Nucleoporin Nup210 Develop Peripheral T Cell Alterations. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Bartosch, B.; Vitelli, A.; Granier, C.; Goujon, C.; Dubuisson, J.; Pascale, S.; Scarselli, E.; Cortese, R.; Nicosia, A.; Cosset, F.L. Cell Entry of Hepatitis C Virus Requires a Set of Co-receptors that Include the CD81 Tetraspanin and the SR-B1 Scavenger Receptor. J. Boil. Chem. 2003, 278, 41624–41630. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Nakagawa, A.; Nagaosa, K.; Shiratsuchi, A.; Nakanishi, Y. Phosphatidylserine binding of class B scavenger receptor type I, a phagocytosis receptor of testicular sertoli cells. J. Boil. Chem. 2002, 277, 27559–27566. [Google Scholar] [CrossRef] [PubMed]
- Witt, W.; Kolleck, I.; Fechner, H.; Sinha, P.; Rüstow, B. Regulation by vitamin E of the scavenger receptor BI in rat liver and HepG2 cells. J. Lipid Res. 2000, 41, 2009–2016. [Google Scholar] [PubMed]
- Hayek, M.G.; Taylor, S.F.; Bender, B.S.; Han, S.N.; Meydani, M.; Smith, D.E.; Eghtesada, S.; Meydani, S.N. Vitamin E Supplementation Decreases Lung Virus Titers in Mice Infected with Influenza. J. Infect. Dis. 1997, 176, 273–276. [Google Scholar] [CrossRef]
- Yang, Y.; Wislez, M.; Fujimoto, N.; Prudkin, L.; Izzo, J.G.; Uno, F.; Ji, L.; Hanna, A.E.; Langley, R.R.; Liu, D.; et al. A selective small molecule inhibitor of c-Met, PHA-665752, reverses lung premalignancy induced by mutant K-ras. Mol. Cancer Ther. 2008, 7, 952–960. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Liao, L.X.; Song, X.M.; Wang, L.C.; Lv, H.N.; Chen, J.F.; Liu, D.; Fu, G.; Zhao, M.B.; Jiang, Y.; Zeng, K.W.; et al. Highly selective inhibition of IMPDH2 provides the basis of antineuroinflammation therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E5986–E5994. [Google Scholar] [CrossRef]
- Morita, M.; Ler, L.W.; Fabian, M.R.; Siddiqui, N.; Mullin, M.; Henderson, V.C.; Alain, T.; Fonseca, B.D.; Karashchuk, G.; Bennett, C.F.; et al. A Novel 4EHP-GIGYF2 Translational Repressor Complex Is Essential for Mammalian Development. Mol. Cell. Boil. 2012, 32, 3585–3593. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Agents Chemother. 2014, 59, 1088–1099. [Google Scholar] [CrossRef]
- Stead, R.L.; Proud, C.G. Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a). FEBS Lett. 2013, 587, 2623–2628. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entrez ID | Gene Name | Virus Interaction | IRG |
|---|---|---|---|
| 3250 | haptoglobin-related protein (HPR) | - | X |
| 23225 | nucleoporin 210 (NUP210) | Nsp4 | X |
| 26505 | cyclin and CBS domain divalent metal cation transport mediator 3 (CNNM3) | - | X |
| 5557 | primase (DNA) subunit 1 (PRIM1) | Nsp1 | X |
| 23367 | La ribonucleoprotein domain family member 1 (LARP1) | N | X |
| 382 | ADP ribosylation factor 6 (ARF6) | Nsp15 | X |
| 2802 | golgin A3 (GOLGA3) | Nsp13 | X |
| 949 | scavenger receptor class B member 1 (SCARB1) | Nsp7 | X |
| 10280 | sigma non-opioid intracellular receptor 1 (SIGMAR1) | Nsp6 | |
| 84767 | tripartite motif-containing 51 (TRIM51) | - | X |
| 3615 | inosine monophosphate dehydrogenase 2 (IMPDH2) | Nsp14 | X |
| 9470 | eukaryotic translation initiation factor 4E family member 2 (EIF4E2) | Nsp2 | |
| 55823 | VPS11, CORVET/HOPS core subunit (VPS11) | ORF3a, ORF8 | X |
| 523 | ATPase H+ transporting V1 subunit A (ATP6V1A) | M | X |
| 2876 | glutathione peroxidase 1 (GPX1) | Nsp5_C145A | X |
| 23181 | disco interacting protein 2 homolog A (DIP2A) | - | X |
| 2150 | F2R like trypsin receptor 1 (F2RL1) | ORF9c | X |
| 5817 | poliovirus receptor (PVR) | ORF8 | X |
| 6731 | signal recognition particle 72 (SRP72) | Nsp8 | X |
| 4276 | MHC class I polypeptide-related sequence A (MICA) | - |
| Indispensable | Neutral | Dispensable | Total | |
|---|---|---|---|---|
| All proteins | 1170 (1169) | 2675 (2658) | 2459 (2454) | 6304 (6281) |
| 18.6% (18.6%) | 42.4% (42.3%) | 39.0% (39.1%) | 100% | |
| Driver proteins | 0 (0) | 810 (799) | 1656 (1664) | 2466 (2463) |
| 0% (0%) | 32.8% (32.4%) | 67.2% (67.6%) | 100% | |
| Virus interacting proteins | 32 (30) | 57 (57) | 59 (61) | 148 (148) |
| 21.6% (20.3%) | 38.5% (38.5%) | 39.8% (41.2%) | 100% |
| Entrez ID | Gene Name | Degree | Betweenness | Classification | IRG |
|---|---|---|---|---|---|
| 1174 | adaptor-related protein complex 1 sigma 1 subunit (AP1S1) | 3 (3) | 34.1 (33.0) | D (N) | |
| 3615 | inosine monophosphate dehydrogenase 2(IMPDH2) | 4 (3) | 4090.0 (65.0) | N (D) | X |
| 6993 | dynein light chain Tctex-type 1(DYNLT1) | 11 (11) | 44,093.7 (43,902.7) | D (N) | X |
| 8480 | ribonucleic acid export 1(RAE1) | 6 (5) | 3863.3 (1076.3) | I (N) | |
| 10280 | sigma non-opioid intracellular receptor 1(SIGMAR1) | 3 (2) | 4094.0 (0.0) | I (N) | |
| 10987 | COP9 signalosome subunit 5(COPS5) | 33 (33) | 30,094.4 (29,968.0) | N (I) | |
| 23225 | nucleoporin 210(NUP210) | 4 (3) | 331.1 (325.9) | N (D) | |
| 64326 | ring finger and WD repeat domain 2(RFWD2) | 3 (3) | 207.1 (206.8) | D (N) | |
| 64837 | kinesin light chain 2(KLC2) | 7 (7) | 463.2 (460.3) | D (N) |
| Critical | Intermittent | Redundant | Total | |
|---|---|---|---|---|
| All proteins | 540 (525) | 3322 (3318) | 2442 (2438) | 6304 (6281) |
| 8.6% (8.4%) | 52.7% (52.8%) | 38.7% (38.8%) | 100% | |
| Driver proteins | 540 (525) | 1926 (1983) | 0 (0) | 2466 (2463) |
| 21.9% (21.3%) | 78.1% (80.5%) | 0% (0%) | 100% | |
| Virus interacting proteins | 0 (8) | 79 (75) | 69 (65) | 148 (148) |
| 0% (5.4%) | 53.4% (50.7%) | 46.6% (32.9%) | 100% |
| Entrez ID | Gene Name | Degree | Betweenness | Classification | IRG |
|---|---|---|---|---|---|
| 949 | scavenger receptor class B member 1(SCARB1) | 5 (4) | 1098.0 (0.0) | I (C) | X |
| 3615 | inosine monophosphate dehydrogenase 2(IMPDH2) | 4 (3) | 4090.0 (65.0) | R (I) | X |
| 5817 | poliovirus receptor (PVR) | 7 (6) | 349.6 (0.0) | I (C) | X |
| 8480 | ribonucleic acid export 1(RAE1) | 6 (5) | 3863.3 (1076.3) | R (I) | X |
| 9470 | eukaryotic translation initiation factor 4E family member 2(EIF4E2) | 4 (3) | 672.1 (0.0) | I (C) | |
| 9662 | centrosomal protein 135(CEP135) | 2 (1) | 46.6 (0.0) | I (C) | X |
| 10280 | sigma non-opioid intracellular receptor 1(SIGMAR1) | 3 (2) | 4094.0 (0.0) | R (C) | |
| 23225 | nucleoporin 210(NUP210) | 4 (3) | 331.1 (325.9) | R (I) | X |
| 26092 | torsin 1A interacting protein 1(TOR1AIP1) | 2 (1) | 41.1 (0.0) | I (C) | X |
| 51552 | RAB14, member RAS oncogene family (RAB14) | 2 (1) | 1.0 (0.0) | I (C) | X |
| 113174 | serum amyloid A like 1(SAAL1) | 2 (1) | 78.2 (0.0) | I (C) |
| Drug (Status) | Structure | Target/Viral Protein | Target Function |
|---|---|---|---|
| Myristic acid (Experimental) |  | PVR/ORF8 | Regulate Natural killer cells, polio virus [27] |
| Sphingosine (Experimental) |  | ||
| Phosphatidyl serine (Approved) |  | SCARB1/Nsp7 | Facilitate cell entry, Hepatitis C [31] |
| Tocopherol/Vitamin E (Approved) |  | ||
| PHA-665752 (Experimental) |  | SCARB1/Nsp7 | Facilitate cell entry, Hepatitis C [31] |
| NUP210/Nsp4 | Transport between nucleus and cytoplasm [32] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ackerman, E.E.; Shoemaker, J.E. Network Controllability-Based Prioritization of Candidates for SARS-CoV-2 Drug Repositioning. Viruses 2020, 12, 1087. https://doi.org/10.3390/v12101087
Ackerman EE, Shoemaker JE. Network Controllability-Based Prioritization of Candidates for SARS-CoV-2 Drug Repositioning. Viruses. 2020; 12(10):1087. https://doi.org/10.3390/v12101087
Chicago/Turabian StyleAckerman, Emily E., and Jason E. Shoemaker. 2020. "Network Controllability-Based Prioritization of Candidates for SARS-CoV-2 Drug Repositioning" Viruses 12, no. 10: 1087. https://doi.org/10.3390/v12101087
APA StyleAckerman, E. E., & Shoemaker, J. E. (2020). Network Controllability-Based Prioritization of Candidates for SARS-CoV-2 Drug Repositioning. Viruses, 12(10), 1087. https://doi.org/10.3390/v12101087