3.1. The sTvbS3-mIgG Receptor Immunoadhesin Protein Can Be Expressed to High Levels and Potently Inhibits ASLV(B) Infection in Cultured Avian Cells
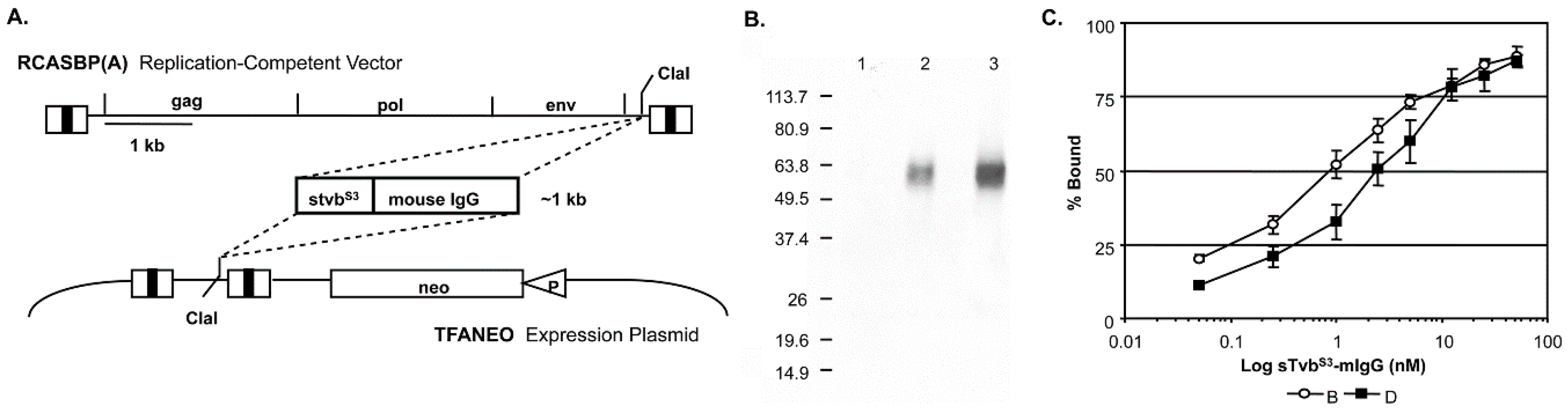
The subgroup A RCASBP(A) retroviral vector that contains and expresses the
stvbS3-mIgG gene used in this study is shown schematically in
Figure 1.
Virus propagation was initiated by transfection of the plasmid containing the retroviral vector into DF-1 cells. The transfected cells were passaged when confluent to allow virus to spread throughout the culture resulting in a chronically infected culture that expressed stable levels of the sTvb
S3-mIgG protein (
Table 1). The same
stvbS3-mIgG gene construct was also subcloned into the expression vector TFANEO, which expresses an experimental gene under the control of the ASLV LTRs and contains the
neo gene under the control of the chicken β-actin promoter (
Figure 1A). Clonal DF-1 cell lines were selected that expressed stable levels of sTvb
S3-mIgG protein. The TF/sTvb
S3-32 cell line expressed the highest level of sTvb
S3-mIgG protein and was used in this study (
Table 1). We did not observe any cytotoxic effects of expressing high levels of the sTvb
S3-mIgG receptor protein in DF-1 cells. This was a concern since high levels of RCASBP(B) replication results in a transient period of cytotoxicity not observed with the RCASBP(A) virus.
The sTvb
S3-mIgG fusion protein has a calculated molecular weight of 41,947 and migrates as a diffuse band (50 to 65 kDa) due to N-linked glycosylation (
Figure 1B). The binding affinities of sTvb
S3-mIgG for subgroup B and D envelope glycoproteins were assayed by FACS using DF-1 cells chronically infected with ASLV(B) or ASLV(D). The sTvb
S3-mIgG receptor protein bound both subgroup B and D glycoproteins with high affinity (
Figure 1C): subgroup B glycoproteins (0.84 ± 0.24 nM) with approximately 3-fold higher affinity compared to subgroup D glycoproteins (2.42 ± 0.55 nM). As expected since Tvb
S3 is specifically the receptor for subgroup B, D and E ASLVs, the sTvb
S3-mIgG protein did not bind to envelope glycoproteins expressed on DF-1 cells infected with ASLV(A) or ASLV(C) at experimentally detectable levels. These estimates of sTvb
S3-mIgG binding affinity may be somewhat high due to the predicted dimer structure of the sTvb
S3-mIgG immunoadhesin.
The level and specificity of the antiviral effect of the sTvb
S3-mIgG receptor protein in cultured cells was quantitated by challenging DF-1 cells expressing sTvb
S3-mIgG from the RCASBP(A) vector, the TF-sTvb
S3-32 stable cell line expressing the sTvb
S3-mIgG from a non-viral expression plasmid, DF-1 cells infected with the RCASBP(A) vector alone, and parental DF-1 cells, with the RCASBP(B)AP or RCASBP(C)AP replication-competent viral vectors. Serial dilutions (10-fold) of the viruses were first incubated with supernatants from confluent control or sTvb
S3-mIgG expressing cultures for 1 h at 4 °C and then plated on the appropriate non-confluent (35% confluent) cells in culture. The infectious titers were determined by assaying for AP activity 2-days after infection. As expected, the sTvb
S3-mIgG receptor protein specifically inhibited entry of subgroup B ASLV but had no significant effect on subgroup C ASLV entry efficiency (
Table 1). In addition, a higher antiviral effect on ASLV(B) entry was observed in the TF/Tvb
S3-32 cells (725-fold inhibition) that expressed the higher level of the sTvb
S3-mIgG receptor protein (40 nM) compared to the RCASBP(A)stvb
S3-mIgG infected cells (70-fold inhibition) that expressed a lower level of sTvb
S3-mIgG (11 nM).
3.2. Delivery, Expression and Antiviral Effect of the sTvbS3-mIgG Immunoadhesin on ASLV(B) Challenge in Chickens
We have previously shown that the RCAS vectors can deliver experimental genes to most tissues in the chicken relatively efficiently by infecting embryos in fertile, unincubated eggs by injecting DF-1 cells producing the replication-competent viral vectors near the embryo (~50,000 cell stage) [
32]. Chicks viremic for the RCAS vector are then identified after hatch by assaying serum for the ASLV CA protein by ELISA. In this study, two groups of viremic chicks were produced: one group infected with the RCASBP(A) vector alone, and a second group infected with RCASBP(A)stvb
S3-mIgG and expressing the sTvb
S3-mIgG receptor protein. There were no observable cytotoxic effects of sTvb
S3-mIgG receptor protein expression in the embryos, hatched chicks or older chickens: hatch rates and survival of birds infected with RCASBP(A) and RCASBP(A)stvb
S3-mIgG were comparable.
The RCASBP(A)-alone infected control group was then divided into three groups: five birds were left unchallenged, 15 birds were challenged with 10
5 ifu RAV-2 (a subgroup B ASLV), and 15 birds challenged with 10
5 ifu RAV-49 (a subgroup C ASLV). The RCASBP(A)stvb
S3-mIgG infected group was divided into two groups: 17 birds were challenged with 10
5 ifu RAV-2, and 22 birds were challenged with 10
5 ifu RAV-49. Blood was collected from each bird eight weeks after challenge, and the serum assayed for subgroups A, B and C ASLV using the In Vitro ASLV Assay (
Table 2).
In the vector alone controls, all of the birds infected with RCASBP(A) alone and still viremic at the end of the experiment, were susceptible to both subgroups B and C ASLV infection and replication. However, viruses were not detected in five birds infected with RCASBP(A) and subsequently challenged with RAV-2 or RAV-49. These birds were technically viremic for ASLV on day 1 after hatch, although expressing relatively low levels of CA compared to the other chicks. We believe that the initial delivery/spread of the virus was not robust enough in these birds to induce immunological tolerance to the ASLV proteins and sustain the viremia once antibody production commenced.
All 22 RCASBP(A)stvbS3-mIgG infected birds challenged with RAV-49 were susceptible to this subgroup C ASLV challenge as expected since the sTvbS3-mIgG protein is specific for subgroup B ASLVs. Only in the RCASBP(A)stvbS3-mIgG infected birds challenged with RAV-2 was an antiviral effect observed: subgroup B ASLV could not be detected in 88% (15/17) of these birds. However, two birds produced detectable RAV-2 in their serum, 6590 and 6620, perhaps coincidentally they were two of the three birds in the group that expressed the lowest levels of sTvbS3-mIgG in their post-hatch serum samples.
3.3. Analysis of the Subgroup B Virus Populations in the Serum of RAV-2 Challenged Birds
Why were birds 6590 and 6620 not completely protected from RAV-2 challenge? Was this the result of incomplete sTvbS3-mIgG gene delivery and/or low expression? Or, do these birds now have mutant RAV-2 virus populations that have escaped the antiviral effect of sTvbS3-mIgG? To answer these questions, we set out to analyze the ASLV(B) populations in the serum of the two birds viremic for ASLV(B), 6590 and 6620, as well as a representative control bird infected with RCASBP(A) vector alone, 6461.
We analyzed the ASLV virus populations in the serum of the birds with both a subgroup A virus, the RCASBP(A) delivery vector, and a subgroup B virus, the RAV-2 challenge virus, by first preferentially amplified subgroup B ASLVs by first passaging the serum in TF/SU(A)-19 cells, a DF-1 cell line expressing SUA-rIgG that is >180,000-fold resistant to ASLV(A) infection. Specifically, ASLVs in 8-week serum samples (100 uL) amplified for three passages (2-days per passage at 1:3 splits) on DF-1 and TF/SU(A)-19 cells [
31]: both subgroup A and B viruses should replicate in DF-1 cells, while subgroup B viruses should preferentially replicate in TF/SU(A)-19 cells. As expected all three birds contained ASLVs that rapidly replicated in DF-1 cells, while only birds 6461, 6590 and 6620 contained virus that replicated well in subgroup A ASLV resistant cells although most likely still contain low levels of subgroup A virus. To test whether mutant subgroup B ASLVs resistant to the antiviral effect of sTvb
S3-mIgG had been selected in these birds, viruses from TF/SU(A)-19 culture supernatants (day 6) were again propagated for three passages in TF/sTvb
S3-32 cells. Only viruses from bird 6620 clearly showed resistance to the antiviral effect of sTvb
S3-mIgG early in the infection (days 2 and 4); however, virus from the other two birds did replicate in these cells, most likely due to residual subgroup A virus. The ASLV
env genes were amplified by PCR from genomic DNA isolated from day 6 infected TF/SU(A)-19 cells, cloned, and the nucleotide sequences of the ASLV
env genes determined. Both subgroup A and subgroup B
env genes were amplified. The complete nucleotide sequence was then determined from 7-10 subgroup B
env gene clones from each bird.
Since only an incomplete nucleotide sequence encoding the RAV-2 SU envelope glycoprotein had been published previously, we initially analyzed subgroup B
env clones from bird 6461 to establish a complete ‘parental’ RAV-2
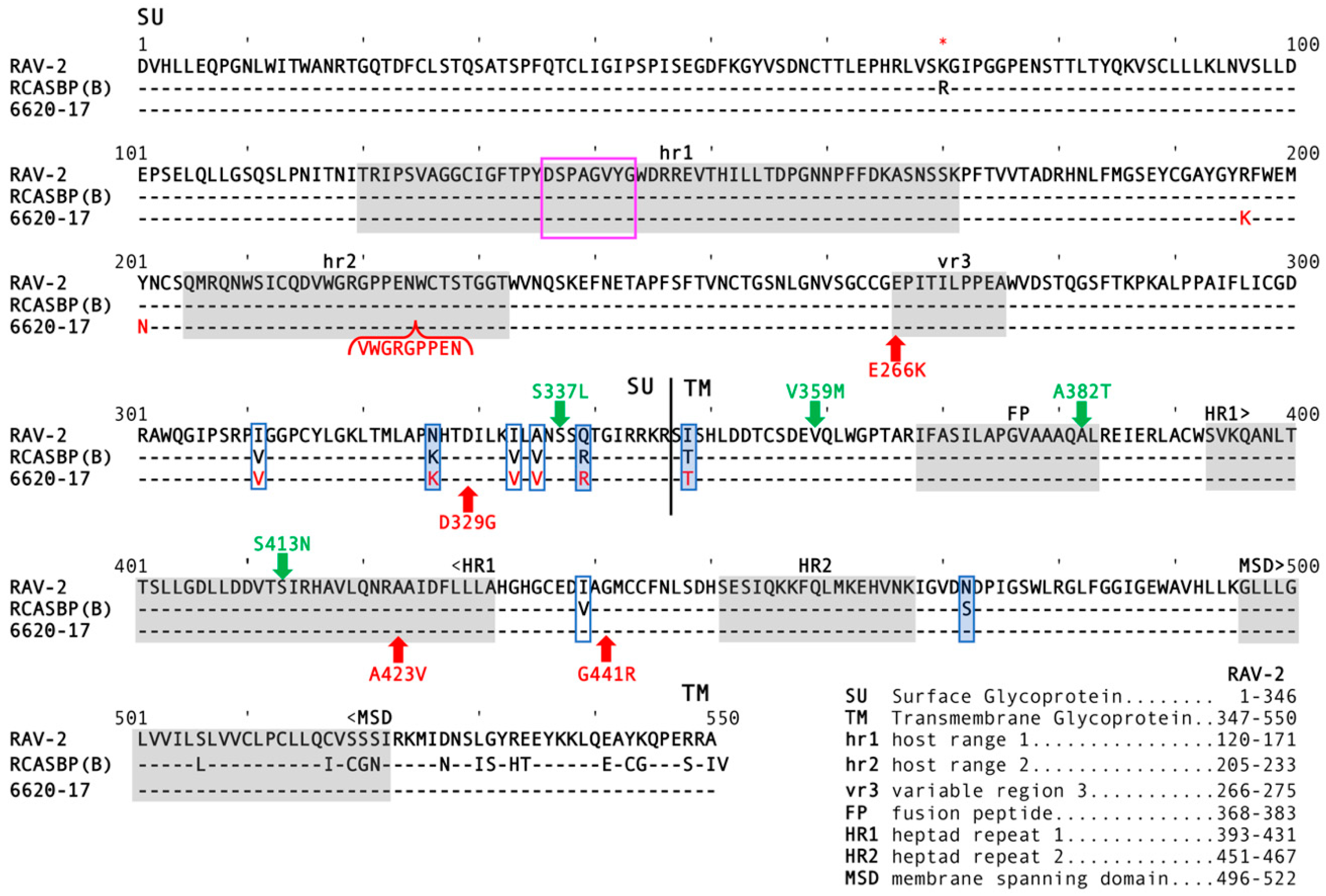
env gene nucleotide sequence. We analyzed seven independent clones (
Figure 2): 4/7 clones contained a RAV-2
env gene; 3/7 clones were obvious recombinants between the
env of RAV-2 and the subgroup A
env of RCASBP(A). The 6461 RAV-2 nucleotide sequence contained 2-3 nucleotide differences compared to the published RAV-2 sequence.
Two of these nucleotide sequence differences were silent changes; however, approximately one-half of the 6461 clones (4/7) and the published RAV-2 sequence coded for arginine (AGA) at amino acid 70 while 3/7 6461 clones coded for lysine (AAA). The amino acid sequence predicted from the 6461 RAV-2
env nucleotide sequence with R70 is shown in
Figure 3 as previously published for RAV-2 (red asterisk). The recombinant RAV-2 and RCASBP(A)
env genes were essentially RAV-2
env genes with ~300 nucleotides of the subgroup A
env gene downstream of the hypervariable regions of SU but upstream of the fusion peptide region of TM (see
Figure 2), a region highly conserved between the different ASLV subgroups. However, there are 6 amino acid differences between RAV-2 and RCASBP(A) in this region (
Figure 3). It is interesting to note that the RCASBP(B) vector includes this region of RCASBP(A) plus the rest of the SR-A TM.
The subgroup B
env genes isolated from bird 6590, infected with RCASBP(A)stvb
S3-mIgG and producing detectable RAV-2 after challenge, were a mixture of “wild-type” RAV-2 genes and similar RAV-2/RCASBP(A) recombinants seen in bird 6461 (
Figure 2). In this bird, all of the “wild-type” RAV-2 genes encoded lysine at position 70 (K70). No other mutations were detected so we concluded that in bird 6590, the delivery and/or expression of the sTvb
S3-mIgG inhibitor was not complete and allowed RAV-2 infection and spread.
3.4. Mutations in Several Different Regions in RAV-2 Env Were Required for Resistance to the sTvbS3-mIgG Antiviral Effect
To test whether any of the subgroup B env genes amplified from bird 6620 coded for envelope glycoproteins resistant to the antiviral effect of sTvbS3-mIgG, RCASBP/AP vectors were constructed with the wild-type RAV-2 env gene, the 6620-17 env gene that contains R196K, Y201N, 27 bp duplication in hr2 and the SU carboxy-terminus region of subgroup A env, the 6620-16 env gene that contains only the SU carboxy-terminus region of subgroup A env, and the 6620-18 env gene that contains only the 27 bp duplication in hr2. TF/sTvbS3-32 cells were transfected with the plasmids encoding viruses with the wild-type RAV-2 env or clone 16, 17 or 18 mutant env and the cells passaged to allow virus replication and spread. Only TF/sTvbS3-32 cells transfected with clone 6620-17 supported virus replication but only at very low levels until after ~15-days when virus replication rapidly increased.
This implied that the 6620-17 virus was at least partially resistant to sTvbS3-mIgG inhibitor, especially in the 6620 bird that expressed much lower levels compared to the TF/sTvbS3-32 cells, but the virus may need additional mutations to efficiently escape the high sTvbS3-mIgG levels in the TF/sTvbS3-32 cells.
TF/sTvb
S3-32 cells were transfected with the plasmids encoding viruses with the wild-type RAV-2
env, 6620-17 env or RCASBP(A) as a positive control. The transfected cells were passaged when confluent to allow virus replication and spread. As expected since the antiviral effect of sTvb
S3-mIgG is specific for subgroups B, D and E ASLVs, the RCASBP(A) control rapidly established a chronically infected culture of TF/sTvb
S3-32 cells. Only the 6620-17 mutant virus but not the parental RAV-2 virus was able to replicate in TF/sTvb
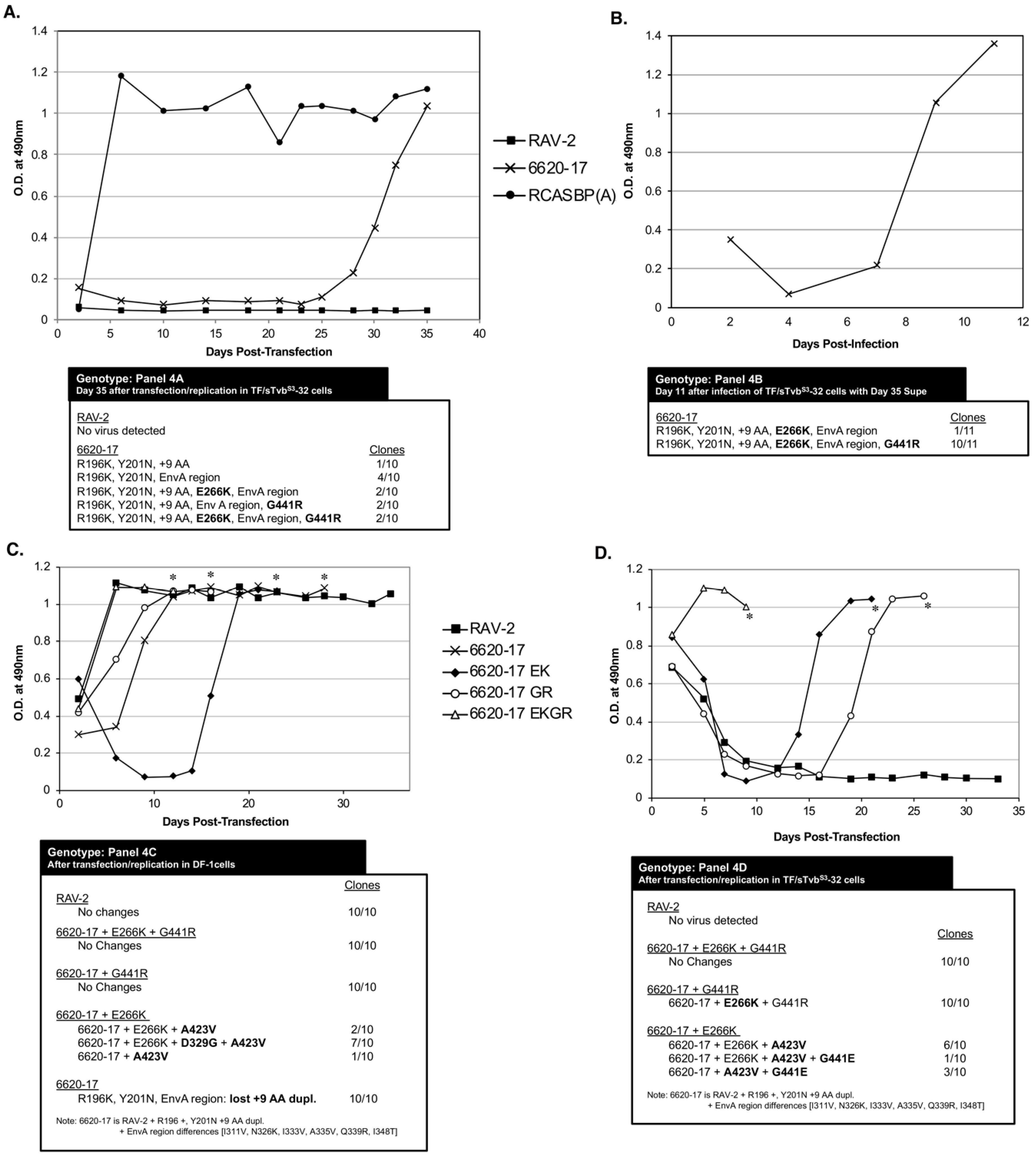
S3-32 cells in the 5-week experiment (
Figure 4A). Only low levels of the 6620-17 virus were detected until day 25 post-transfection when a significant increase in virus production was observed. The significant increase in the production of 6620-17 virus after 3-weeks of replication in TF/sTvb
S3-32 cells implies that the virus may have evolved to increase its resistance to the high concentration of sTvb
S3-mIgG by acquiring additional mutations in the
env gene. To test if an escape mutant virus population was selected, day 35 cell culture supernatant (100 ul) from 6620-17 infected TF/sTvb
S3-32 cells was used to infect fresh TF/sTvb
S3-32 cells, and the infected cells passaged when confluent to allow virus replication and spread (
Figure 4B). This 6620-17 virus population rapidly replicated in fresh TF/sTvb
S3-32 cells without a lag period further implying that additional mutations were acquired that improve resistance to sTvb
S3-mIgG.
The ASLV
env genes were amplified by PCR from genomic DNA isolated from day 35 TF/sTvb
S3-32 cells transfected with 6620-17 (
Figure 4A) and day 11 TF/sTvb
S3-32 cells infected with day 35 6620-17 supernatant (
Figure 4B), and cloned. The nucleotide sequences of the ASLV
env genes were determined from at least ten individual clones from each culture, and the corresponding amino acid sequences compared to the original 6620-17 mutant protein encoded by the plasmid used to initiate infection. The virus population produced with the original 6620-17 Env sequence did evolve after prolonged initial passage in TF/sTvbS3-32 cells to a population dominated (10/11 clones) with the 6620-17 Env but with two new, additional mutations: E266K mutation in the SU vr3 variable region, and the G441R mutation in the critical chain reversal region between the two heptad repeats HR1 and HR2 in the TM glycoprotein.
To characterize these new mutations, three new RCASBP plasmids were constructed containing the 6620-17
env gene adding the E266K mutation (EK), or 6620-17 adding the G441R (GR) mutation or adding both mutations (EKGR) in the 6620-17 Env background. These three plasmids plus the control RAV-2 plasmid were transfected into normal DF-1 cells to assess viral fitness (
Figure 4C). Both 6620-17 + EKGR and 6620-17 + g and the control RAV-2 viruses replicated well in DF-1 cells without significant time lags in reaching maximum virus levels, with no additional genomic changes were found in these viruses. The 6620-17 parent virus replicated with a slight delay in reaching maximum levels and the genomes of 10/10 clones had all lost the 9 amino acid duplication in SU hr2 region leading to the possible conclusion that without the sTvb
S3-IgG inhibitor, the 9 AA duplication is not needed and its deletion results in a more fit virus. Finally, the replication of the 6620-17+EK virus required a significant delay before virus replication reach high levels. The resulting virus pool had several genotypes with the majority of genomes (7/10) acquiring two mutations: the D320G mutation in the C-terminal region of SU and the A423V mutation in the HR1 region. The E266K mutation knocks out the ability of the 6620-17 + EK and the 6620-17 + EKGR Env glycoproteins to detectably bind to the sTvb
S3 receptor while 6620-17+GR and RAV-2 bind the sTvb
S3 receptor with wild-type affinities.
The 6620-17, 6620-17+EK, 6620-17+GR and 6620-17 + EKGR plasmids plus the control RAV-2 plasmid were also transfected into TF/sTvbS3-32 cells to assess viral resistance to the sTvb
S3-IgG inhibitor (
Figure 4D). Only the 6620-17 + EKGR virus containing both the E266K mutation that knocks out the ability of the sTvb
S3-IgG inhibitor to bind the ASLV(B) Env, and the G441R mutation in the chain reversal region of the TM, replicated efficiently in the presence of the sTvb
S3-IgG in TF/sTvb
S3-32 cells. Both of the 6620-17 viruses with single mutations required a significant time delay before either virus replicated efficiently with 10/10 clones of the 6620-17 + GR virus acquiring the E266K mutation, and the 6620-17 + EK virus acquiring one or more mutations in TM, A423V and or G441E. Again, as observed in the experiment of
Figure 4A, a detectable level of the wild-type RAV-2 virus was again never produced after transfection of TF/sTvbS3-32 cells.
3.5. Residues in the C-Terminal End of SU and in TM in RAV-2 Alter the Biophysical Properties of Virus Entry
The subgroup B RCASBP(B) vector was originally constructed by replacing just the N-terminal region of SR-A RCASBP(A) Env with the complementary region of RAV-2 SU. At the time, only the nucleotide sequence of this region of RAV-2 Env had been determined and cloned, and this ASLV Env region of SU had been shown to confer receptor specificity. Therefore, RCASBP(B) Env is composed of the first ~280 residues of RAV-2 SU fused to the SR-A Env (see
Figure 3 for comparison to the complete RAV-2 Env determined in this study). The extracellular amino acid sequence is highly conserved in the C-terminal of the ASLV SU hypervariable regions (
Figure 3), although there are 8 extracellular amino acid differences between RAV-2 and SR-A in this region. The region acquired by the 6620-17 virus from the SR-A RCASBP(A) vector contained 6/8 extracellular amino acid Env differences between SR-A and RAV-2 (
Figure 3).
Since both viruses replicate at approximately the same rate and to the same titers in DF-1 cells, we hypothesized that the amino acid differences between RAV-2 and RCASBP(B) altered the viruses fundamental ability to promote virus entry in some way, likely altering the two-step mechanism of ASLV Env fusion process for entry. The conformation of the mature, metastable RAV-2 and RCASBP(B) glycoprotein trimers on wild-type virions purified from DF-1 cells were analyzed using the TM oligomerization assay conditions based on experimental conditions defined by Smith et al. [
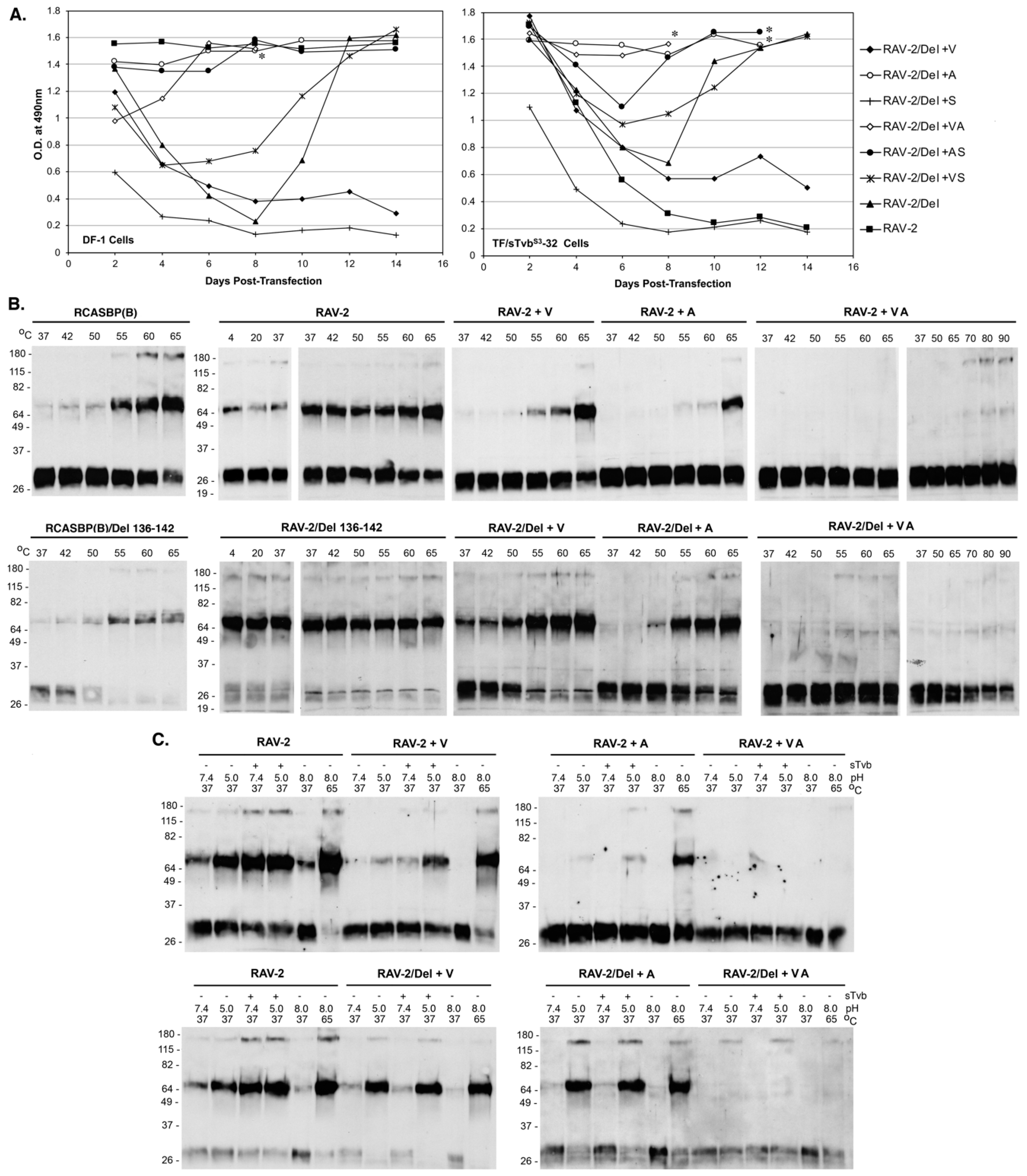
25]. The heat denaturation of RCASBP(B) glycoprotein trimers triggered the trimers to form stable, SDS-resistant TM oligomers initially at ~55 °C, but predominately at 60–65 °C, with almost all TM in oligomers at 65 °C: TM monomers (~30 kDa) form two oligomeric species of 70–80 kDa and ~170 kDa (
Figure 5A). The heat denaturation of RAV-2 glycoprotein trimers also formed TM oligomers at predominately 60–65 °C, but a low but significant level of the 70–80 kDa TM oligomer was evident at all temperatures tested, as low as 37 °C, indicating the RAV-2 glycoprotein trimer structure was not as stable compared to the RCASBP(B) glycoprotein trimers.
The combination of receptor-induced conformation changes and a subsequent low pH exposure of RCASBP(B) envelope glycoprotein trimers at 37 °C produced TM oligomers as expected: the combination of first receptor binding at 37 °C to triggering structural changes in SU, followed by low pH exposure (pH 5.0) efficiently produced the highest levels of TM oligomers (
Figure 5B). In contrast, the RAV-2 glycoprotein trimers again did not demonstrate the same level of specificity as RCASBP(B) glycoprotein trimers for receptor binding followed by low pH exposure to efficiently produce stable TM oligomers. Again, a low level of TM oligomers was observed at 37 °C without receptor binding and at neutral pH 7.4, and pH 5.0 exposure did trigger a higher level of TM oligomers without receptor binding. However, RAV-2 glycoprotein trimers upon binding receptor do not appear to require a low pH exposure to produce maximum levels of stable TM oligomers since exposure to pH 7.4 and pH 5.0 resulted in similar TM oligomer levels.
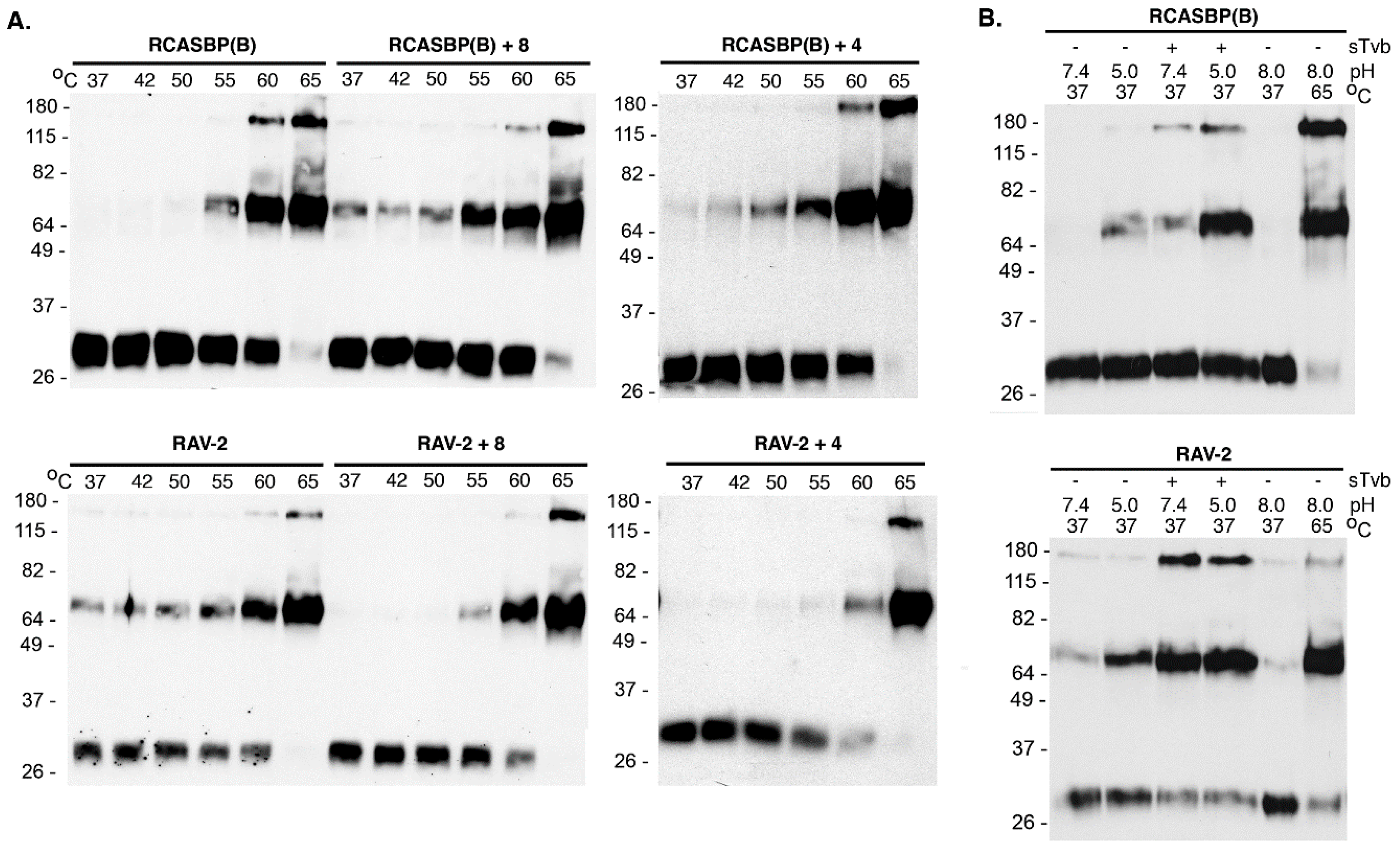
To determine if the 8-extracellular amino acid differences between RCASBP(B) and RAV-2 (see
Figure 3) were responsible for their different biophysical properties, the SR-A Env glycoprotein residues of RCASBP(B) were converted to RAV-2 residues generating RCASBP(B) + 8, and the reverse for RAV-2 generating RAV-2 + 8. DF-1 cells were transfected with these plasmids, cultured, and virions harvested and purified and the properties of their metastable glycoprotein trimers analyzed using heat denaturation TM oligomerization assays. Changing these 8-residues reversed the characteristic heat denaturation pattern of TM oligomer formation to the virus providing the 8-amino acid residues: the assay result of RCASBP(B) + 8 was now identical to RAV-2; while the assay result of RAV-2 + 8 was identical to RCASBP(B) (
Figure 5A). Since four of the eight amino acid differences are conservative amino acid substitutions, two new viruses were constructed that just changed the four non-conserved differences to generate RCASBP(B) + 4 and RAV-2 + 4 viruses. Similar results were obtained with just these four non-conserved amino acid changes: the heat denaturation TM oligomer assay result of RCASBP(B)+4 was now identical to RAV-2; while the assay result of RAV-2 + 4 was identical to RCASBP(B). The combination receptor-induced conformation changes and a subsequent low pH exposure of purified RCASBP(B)+4 and RAV-2 + 4 viruses at 37 °C also reversed the assay results where RCASBP(B) + 4 was now identical to RAV-2; while the assay result of RAV-2 + 4 was identical to RCASBP(B) (data not shown).
These data imply these four amino acid differences produce structural changes in these regions of the SU and TM glycoproteins that alter the interplay between SU and TM glycoproteins in the Env trimer that alter the efficiency of virus entry and ability to adapt/evolve their receptor usage to escape the sTvbS3-mIgG inhibitor.
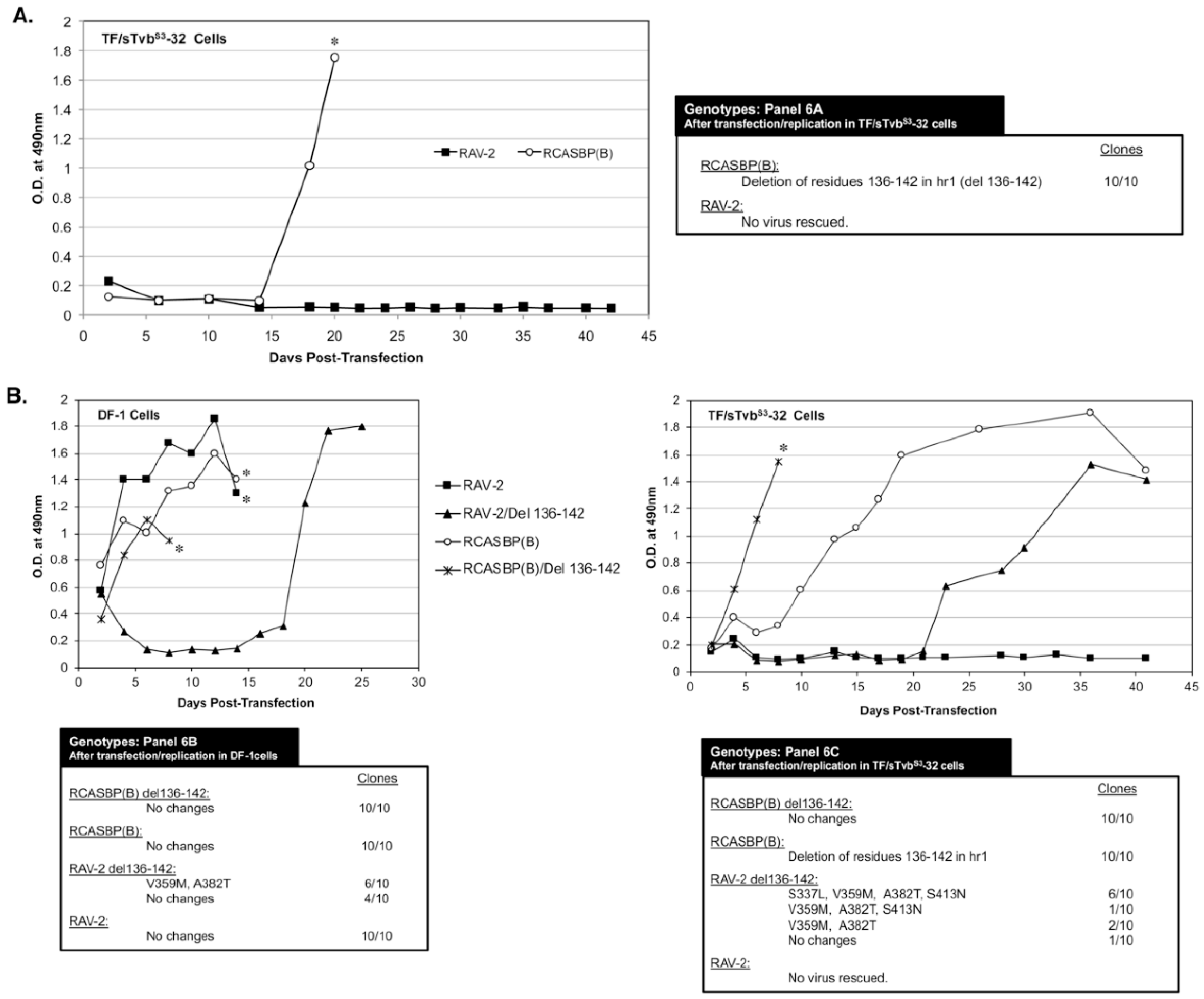
3.6. The 136-142 hr1 Deletion in RCASBP(B) Env That Rescued the RCASBP(B) Virus from the sTvbS3-IgG Inhibitor Does Not Rescue RAV-2
To characterize the possible effects of amino acid differences in this region on the ability of ASLV to escape the antiviral effect of sTvb
S3-IgG, TF/sTvbS3-32 cells were transfected with RAV-2 (RCASBP containing the entire RAV-2
env gene) and RCASBP(B) plasmids and passaged to select for possible escape variants. After a lag period of ~15-days, rapidly replicating virus was detected in the RCASBP(B) transfected culture (
Figure 6A). However, as was observed in the previous two experiments, no virus was produced in the cells transfected with RAV-2. The RCASBP(B)
env genes cloned from the apparent escape virus pool all contained the same deletion, deleting residues 136-142 in the hr1 region of SU. Both RAV-2 and RCASBP(B) contain this same SU variable regions but only viable escape mutations occurred in the RCASBP(B) virus background. An RCASBP(B) virus was constructed with the 136-142 hr1 deletion, RCASBP(B)/Del136-142: this deletion knocks out the ability of the RCASBP(B)/Del136-142 Env glycoproteins to detectably bind to the sTvb
S3 receptor while RCASBP(B) Env glycoproteins bind the sTvb
S3 receptor with wild-type affinity.
Since multiple attempts have failed to rescue a RAV-2 based virus in the presence of the sTvb
S3-IgG inhibitor using TF/sTvbS3-32 cells, we constructed RAV-2 with this same hr1 136-142 deletion, RAV-2/Del136-142. The four plasmids encoding the wild-type and deletion mutant viruses were transfected into DF-1 cells and TF/sTvbS3-32 cells, passaged, and the subsequent viruses produced analyzed (
Figure 6B). RCASBP(B) and RCASBP(B)/Del136-142 replicated well in DF-1 cells without production lags and without new Env mutations being selected. As expected, RCASBP(B)/Del136-142 replicated well in the presence of the sTvb
S3-IgG inhibitor in TF/sTvbS3-32 cells without additional Env mutations, while a lag in the production of virus was observed in parent RCASBP(B) culture with selection of the same 136-142 deletion in the escaped viruses as the previous experiment.
The parental RAV-2 virus replicated well in DF-1 cells without genomic changes, and as in previous experiments, RAV-2 did not replicate in the presence of the sTvb
S3-IgG inhibitor in TF/sTvbS3-32 cells, and no escape RAV-2 virus variants were selected (
Figure 6B). Unexpectedly, the constructed RAV-2/Del136-142 virus did not replicate well even in DF-1, with virus production lagging for ~15-days before a significant replication improvement indicating a selection of possible new glycoprotein mutations. The RAV-2/Del136-142 virus pool from DF-1 cells contained a mixture of the parental RAV-2/Del136-142 virus, and viruses with two additional mutations, V359M located in the N-terminal end of the TM glycoprotein, and A382T located in the fusion peptide (
Figure 3). In the presence of the sTvb
S3-IgG inhibitor in TF/sTvbS3-32 cells, the hr1 deletion in the constructed RAV-2/Del136-142 did not directly rescue virus propagation, but a pool of escape virus was selected that retained the Del136-142 deletion and predominately added four mutations: S337L in the C-terminal end of SU, the same V359M and A382T TM mutations, and an additional mutation S413N located in the heptad repeat 1 (HR1) region of TM. This was the first instance that an RAV-2 based virus escaped the antiviral effects of the sTvb
S3-IgG inhibitor but required the hr1 deletion as a starting point for evolution.
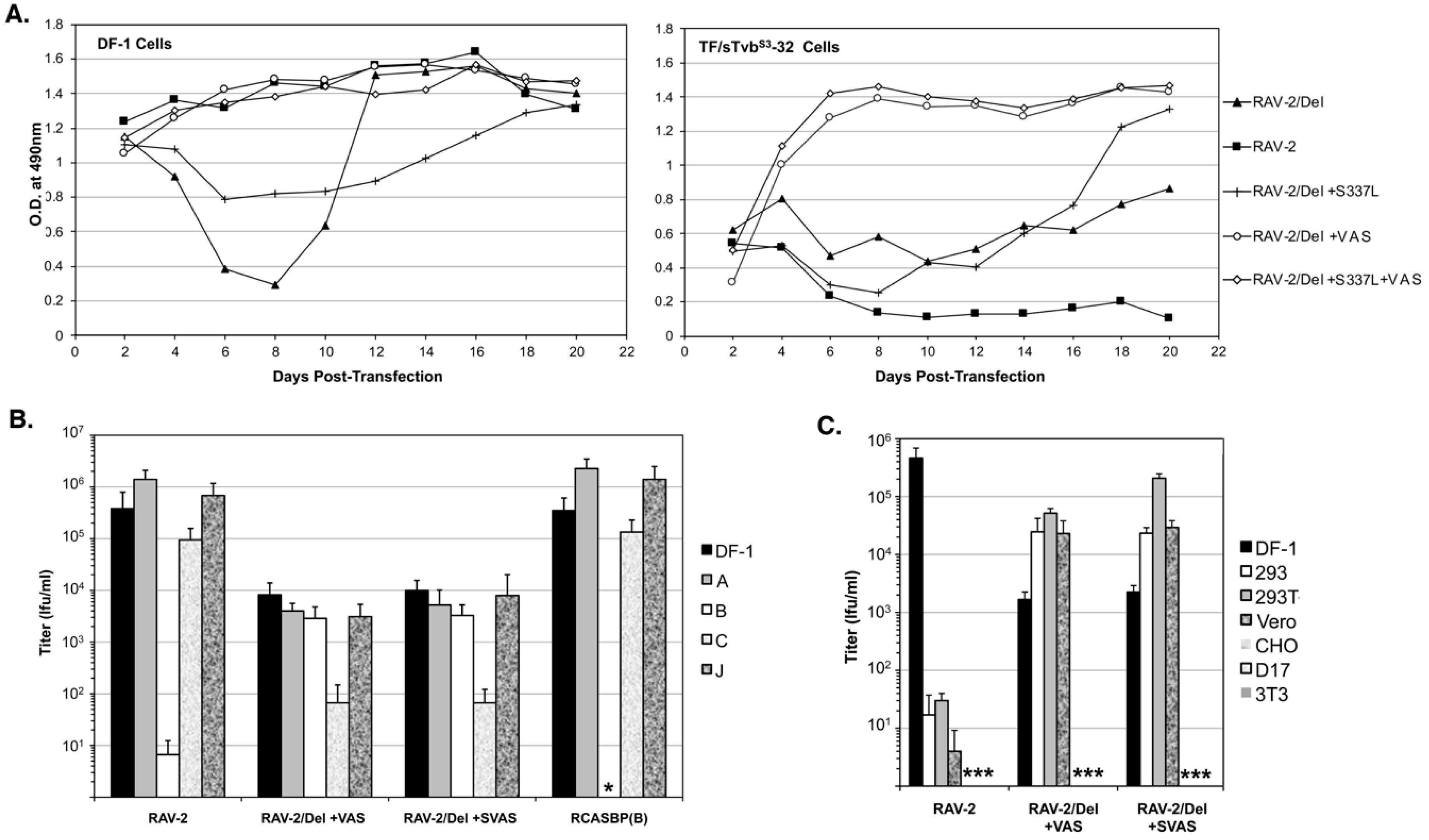
3.7. The Selected RAV-2/Del136-142 Escape Mutants Have Altered Receptor and Host Range Specificities
To determine if all the selected mutations are required for efficient RAV-2/Del136-142 virus replication, the plasmid encoding RAV-2/Del136-142 virus was engineered with all four selected mutations, S337L + VAS, the three mutations in TM, +VAS, and only the SU mutation S337L, and the four plasmids transfected into DF-1 and TF/sTvbS3-32 cells (
Figure 7A). Both the RAV-2/Del136-142 virus with all four mutations +S337L + VAS and only the three TM mutations + VAS, replicated well in both DF-1 and TF/sTvbS3-32 cells. The SU S337L mutation when present alone in RAV-2/Del136-142 does not improve replication in either DF-1 or TF/sTvbS3-32 cells.
The titers of the RAV-2, RAV-2/Del+VAS and RAV-2/Del+SVAS viruses produced by the transfected DF-1 cells that did not appear to lag in replication (see
Figure 7A) were determined using a receptor interference assay using uninfected DF-1 cells and DF-1 cell cultures previously infected with RCASBP(A), RCASBP(B), RCASBP(C) or a subgroup J ASLV. The RAV-2 and RCASBP(B) controls demonstrate the expected receptor interference assay result of a subgroup B ASLV only being blocked by DF-1 cells previously infected by another subgroup B ASLV (
Figure 7B).
However, both the RAV-2/Del+VAS and RAV-2/Del+SVAS viruses can infect DF-1/B cells as efficiently as uninfected DF-1 cells, and both viruses are interfered with the infection of DF-1/C cells where the Tvc receptor would be blocked, producing ~20-fold lower titers compared to the other cells. A panel of mammalian cell lines and DF-1 cells were used to determine if the host range of the mutant viruses had also been altered. The RAV-2 control demonstrates the usual ASLV(B) host range restriction of ASLV to avian cells since mammalian cells do not express ASLV receptors, a 4-log reduction in infection efficiency (
Figure 7C). However, the +VAS mutations in the TM glycoprotein enable both the RAV-2/Del + VAS and RAV-2/Del + SVAS viruses to infect several mammalian cell lines at levels >10-fold higher compared to the viruses ability to infect DF-1 cells.
3.8. The Selected Mutations in the RAV-2/Del136-142 TM Glycoprotein Alter the Biophysical Properties of the RAV-2/Del136-142 Env Glycoprotein Trimer to Allow Efficient Entry, Evasion of the sTvbS3-IgG Inhibitor, and Productive Virus Replication
To determine the minimum mutations required for rescue of RAV-2/Del136-142 virus replication, viruses were generated with combinations of single and double mutations of the three VAS TM mutations, and the plasmids transfected into DF-1 and TF/sTvbS3-32 cells (
Figure 8A). Only the RAV-2/Del + VA and RAV-2/Del + A viruses replicated relatively well in both DF-1 and TF/sTvbS3-32 cells without significant lags in virus production. The V359M and A382T mutations appear to be the minimum changes required for recovering relatively efficient RAV-2/Del136-142 virus replication.
We hypothesized that the escape mutations selected in TM altered the metastable and triggering properties of the RAV-2/Del136-142 glycoprotein trimers to provide an advantage to increase the efficiency of viral entry and subsequent propagation. The same mutations were also constructed in the RAV-2 virus background to separate the possible effects from the Del136-142 mutation. To characterize the biophysical properties of the viral glycoprotein trimers, TM oligomerization assays were done with virions purified from 2-day transient transfections to ensure that the predominant virus species would be encoded by the plasmid and before any possible selection of additional mutations (
Figure 8B). In general, we observe a slight difference in the results of the temperature denaturation TM oligomerization assays when using virus purified from transient transfections: slightly more ‘background’ TM oligomers are observed at lower temperatures; and a relative increase in the TM oligomer levels at 55 °C for RCASBP(B) (compare RACSBP(B) in
Figure 8B to
Figure 5A). The temperature denaturation of RCASBP(B) and RCASBP(B)/Del136-142 produced a similar pattern of TM oligomers as would be expected since the Del136-142 likely predominately only alters sTvb
S3-IgG receptor binding but not stability of the fundamental trimer structure. For RAV-2 virions purified from transient transfections, similar levels of TM oligomers are produced at all temperatures (compare RAV-2 in
Figure 8B to
Figure 5A). For RAV-2/Del136-142, the trimer does appear to form higher levels of TM oligomers at all temperatures compared to RAV-2 indicating the Del136-142 deletion does alter the stability of the trimer and may explain the lack of initial replication in DF-1 cells.
As hypothesized, the mutations acquired in the TM glycoprotein of RAV-2/Del136-142 altered the stability and triggering properties of the both the RAV-2 parental and the RAV-2/Del136-142 Env glycoprotein trimers. The single V359M mutation (+V) and the single A382T (+A) mutation altered the heat denaturation TM oligomer assay results of both the RAV-2/Del136-142 and RAV-2 Env trimers to resemble the RCASBP(B)/Del136-142 and RCASBP(B) Env trimer results upon heat denaturation although RAV-2 + A appears to produce lower levels of TM oligomers compared to RAV-2 + V (
Figure 8B). However, both RAV-2/Del136-142 and RAV-2 Env trimers that contain both the V350M and A382T mutations (+VA) do not form high levels of TM oligomers at any temperature even up to 90 °C. RAV-2/Del136-142 and RAV-2 Env trimers that contain the three TM mutations (+VAS) and all four mutations (+SVAS) also do not produce significant levels of TM oligomers even up to 90 °C and look similar to the +VA results.
The TM mutations also altered the RAV-2 and RAV-2/Del136-142 responses to Env trimer triggering by receptor binding and exposure to low pH (
Figure 8C). The single V359M mutation altered RAV-2 Env trimer TM oligomer formation to specifically require sTvb receptor binding and pH 5.0 exposure to optimally trigger TM oligomer formation in a pattern very similar to RCASBP(B) (compare RAV-2 + V
Figure 8C to RCASBP(B)
Figure 5B). As with the V359M mutation, the single A382T mutation also altered RAV-2 Env trimer TM oligomer formation to require both receptor binding and low pH exposure (RAV-2 + A), but the overall levels of TM oligomer formed were much lower compared to RAV-2+V and RCASBP(B). The V359M mutation and A382T single mutations in the RAV-2/Del136-142 background also altered the receptor pH TM oligomer formation, but in this case the RAV-2/Del+V Env trimer and RAV-2/Del + A was only triggered to form TM oligomers by exposure to pH 5.0 without first requiring receptor binding and SU structural changes. As was observed in the heat denaturation TM oligomer assays, both RAV-2/Del136-142 and RAV-2 Env trimers that contain both the V350M and A382T mutations (+VA) do not form high levels of TM oligomers under any experimental condition of receptor binding and low pH exposure. RAV-2/Del136-142 and RAV-2 Env trimers that contain the three TM mutations (+VAS) and all four mutations (+SVAS) also do not appear to produce high levels of TM oligomers and look similar to the +VA results.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
