Attenuation of Bluetongue Virus (BTV) in an in ovo Model Is Related to the Changes of Viral Genetic Diversity of Cell-Culture Passaged BTV

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Lines and Viruses
2.3. Infection Studies in Chicken Embryos
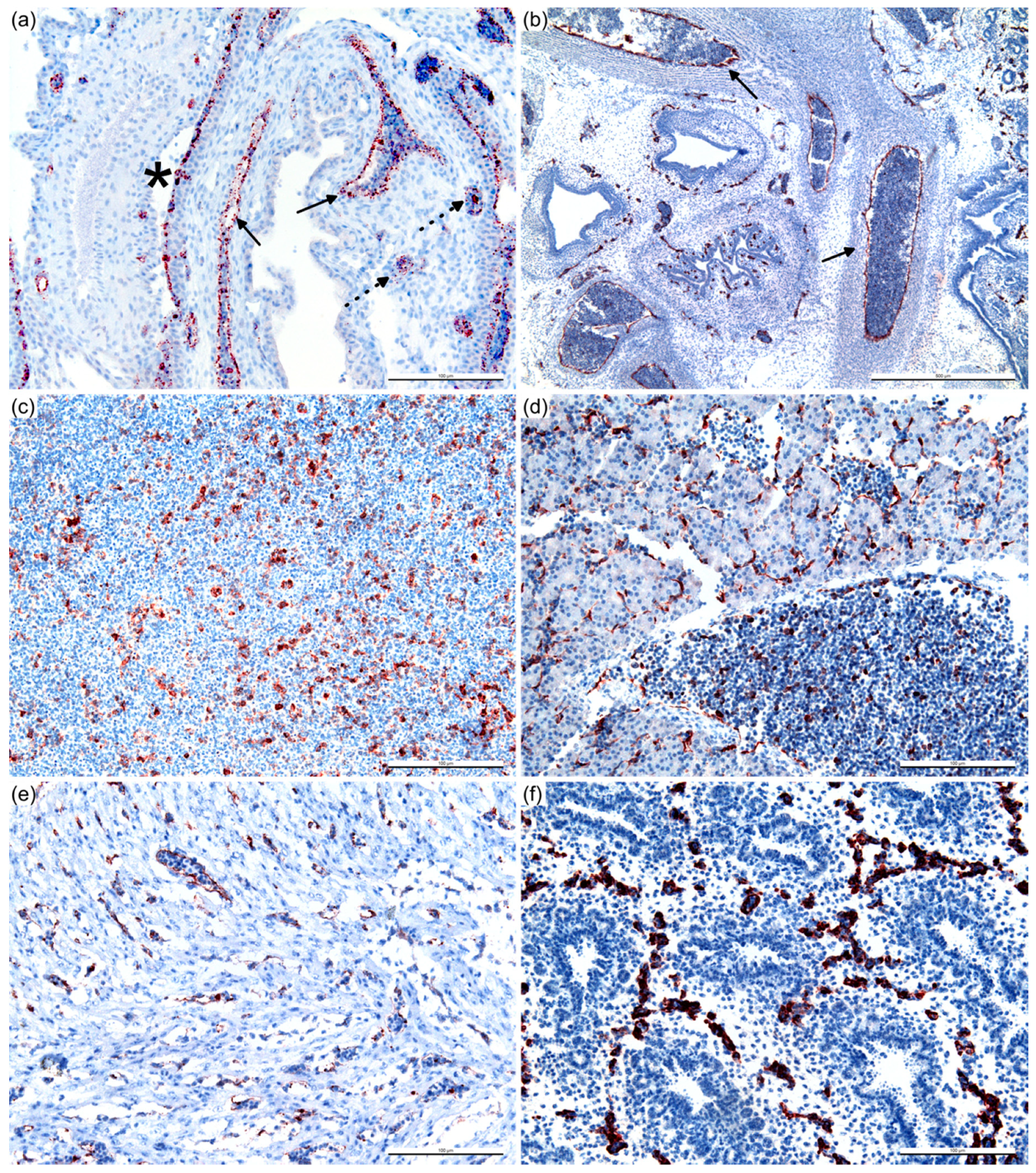
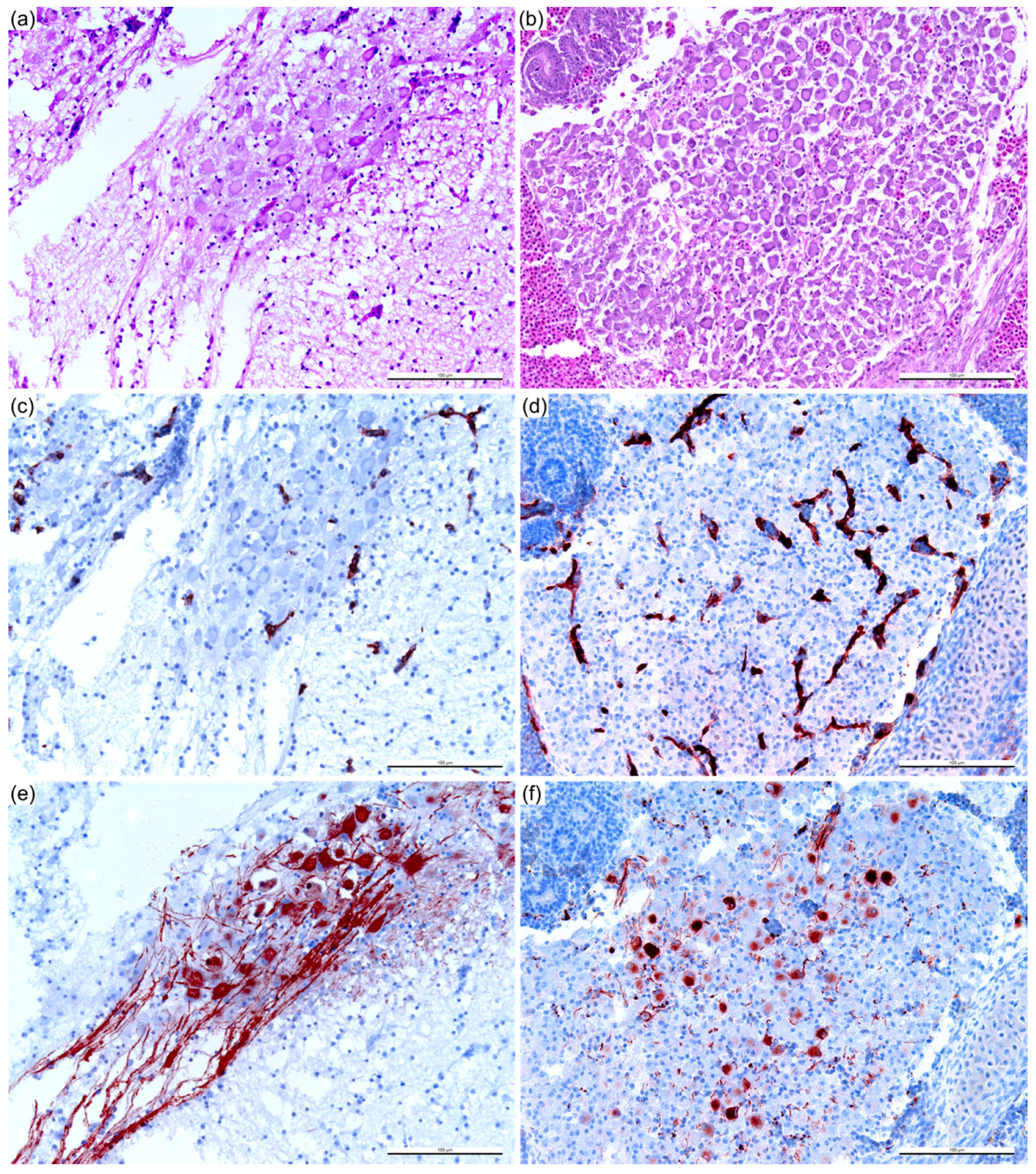
2.4. Histopathology and Immunohistochemistry (IHC)
2.5. Assessment of the Immunohistochemical Labeling
2.6. One-Step Growth Curve Analysis
2.7. Deep Sequencing
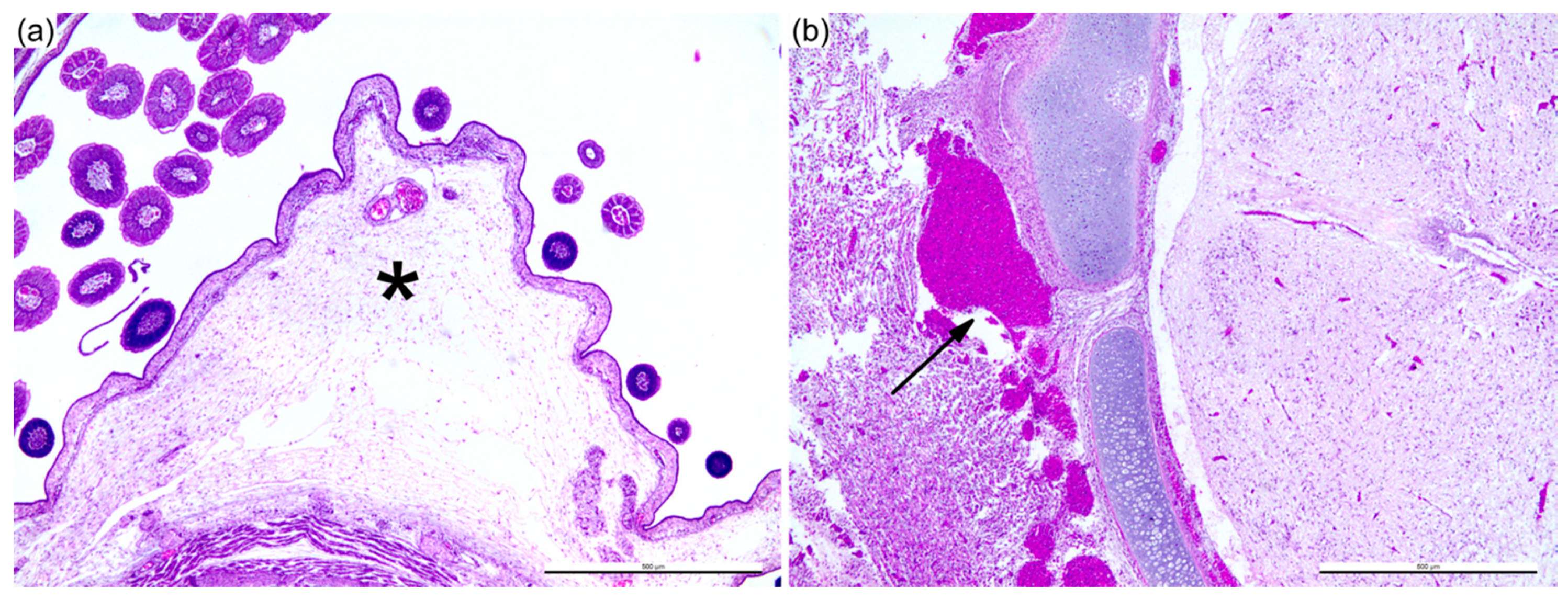
3. Results
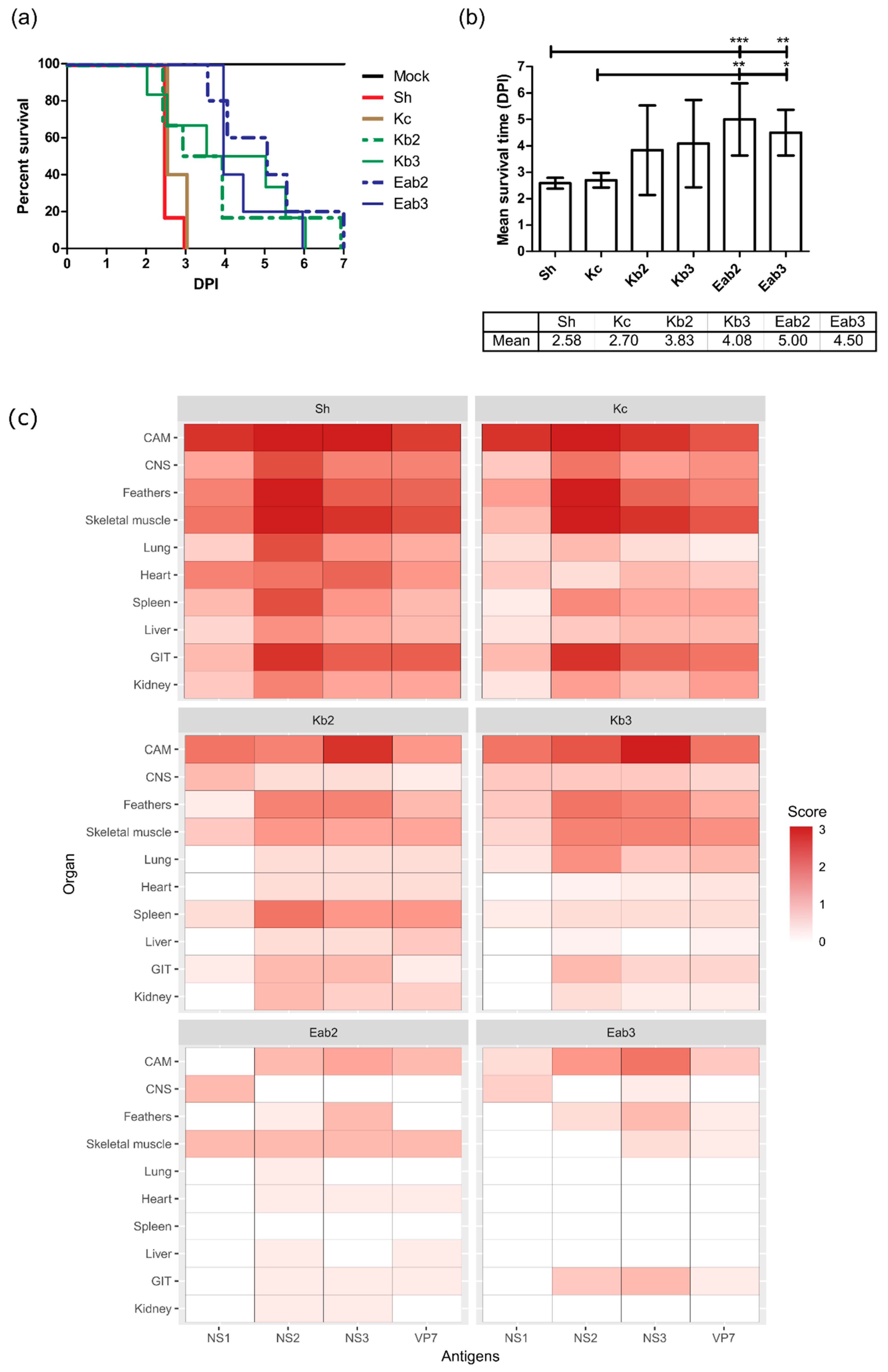
3.1. Sheep Blood Containing BTV-3 is Pathogenic in the ECE Model
3.2. BTV Propagation Method Affects Replicative Fitness In Vitro
3.3. Reduction of Pathogenicity In Ovo with Cell-Culture-Passaged BTV
3.4. Gene Segment-Specific Mutations and Virus Diversity within BTV Populations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maclachlan, N.J.; Zientara, S.; Wilson, W.C.; Richt, J.A.; Savini, G. Bluetongue and epizootic hemorrhagic disease viruses: Recent developments with these globally re-emerging arboviral infections of ruminants. Curr. Opin. Virol. 2019, 34, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Ooi, E.-E.; Horstick, O.; Wills, B. Dengue. The Lancet 2019, 393, 350–363. [Google Scholar] [CrossRef]
- Linthicum, K.J.; Britch, S.C.; Anyamba, A. Rift valley fever: An emerging mosquito-borne disease. Annu. Rev. Entomol. 2016, 61, 395–415. [Google Scholar] [CrossRef]
- Baud, D.; Gubler, D.J.; Schaub, B.; Lanteri, M.C.; Musso, D. An update on zika virus infection. Lancet 2017, 390, 2099–2109. [Google Scholar] [CrossRef]
- Varela, M.; Schnettler, E.; Caporale, M.; Murgia, C.; Barry, G.; McFarlane, M.; McGregor, E.; Piras, I.M.; Shaw, A.; Lamm, C.; et al. Schmallenberg virus pathogenesis, tropism and interaction with the innate immune system of the host. PLoS Pathog. 2013, 9, e1003133. [Google Scholar] [CrossRef]
- Maclachlan, N.J.; Osburn, B.I. Teratogenic bluetongue and related orbivirus infections in pregnant ruminant livestock: Timing and pathogen genetics are critical. Curr. Opin. Virol. 2017, 27, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Caporale, M.; Di Gialleonorado, L.; Janowicz, A.; Wilkie, G.; Shaw, A.; Savini, G.; Van Rijn, P.A.; Mertens, P.; Di Ventura, M.; Palmarini, M. Virus and host factors affecting the clinical outcome of bluetongue virus infection. J. Virol. 2014, 88, 10399–10411. [Google Scholar] [CrossRef]
- Calvo-Pinilla, E.; Rodriguez-Calvo, T.; Anguita, J.; Sevilla, N.; Ortego, J. Establishment of a bluetongue virus infection model in mice that are deficient in the alpha/beta interferon receptor. PLoS ONE 2009, 4, e5171. [Google Scholar] [CrossRef] [PubMed]
- Goldsmit, L.; Barzilai, E. An improved method for the isolation and identification of bluetongue virus by intravenous inoculation of embryonating chicken eggs. J. Comp. Pathol. 1968, 78, 477–487. [Google Scholar] [CrossRef]
- O.I.E. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2018. Available online: http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/3.01.03_BLUETONGUE.pdf (accessed on 2 April 2019).
- Maartens, L.H.; Erasmus, B.J.; Clift, S.J. Tissue tropism of african horsesickness virus in the chicken embryo demonstrated with the avidin-biotin complex immunoperoxidase method. Vet. Pathol. 2011, 48, 1085–1093. [Google Scholar] [CrossRef]
- Wang, L.; Kemp, M.C.; Roy, P.; Collisson, E.W. Tissue tropism and target cells of bluetongue virus in the chicken embryo. J. Virol. 1988, 62, 887–893. [Google Scholar] [PubMed]
- Melzi, E.; Caporale, M.; Rocchi, M.; Martin, V.; Gamino, V.; di Provvido, A.; Marruchella, G.; Entrican, G.; Sevilla, N.; Palmarini, M. Follicular dendritic cell disruption as a novel mechanism of virus-induced immunosuppression. PNAS 2016, 113, E6238–E6247. [Google Scholar] [CrossRef]
- Hamers, C.; Galleau, S.; Chery, R.; Blanchet, M.; Besancon, L.; Cariou, C.; Werle-Lapostolle, B.; Hudelet, P.; Goutebroze, S. Use of inactivated bluetongue virus serotype 8 vaccine against virulent challenge in sheep and cattle. Vet. Record 2009, 165, 369–373. [Google Scholar] [CrossRef]
- Koumbati, M.; Mangana, O.; Nomikou, K.; Mellor, P.S.; Papadopoulos, O. Duration of bluetongue viraemia and serological responses in experimentally infected european breeds of sheep and goats. Vet. Microbiol. 1999, 64, 277–285. [Google Scholar] [CrossRef]
- Moulin, V.; Noordegraaf, C.V.; Makoschey, B.; van der Sluijs, M.; Veronesi, E.; Darpel, K.; Mertens, P.P.; de Smit, H. Clinical disease in sheep caused by bluetongue virus serotype 8, and prevention by an inactivated vaccine. Vaccine 2012, 30, 2228–2235. [Google Scholar] [CrossRef] [PubMed]
- Van Gennip, R.G.; van de Water, S.G.; Maris-Veldhuis, M.; van Rijn, P.A. Bluetongue viruses based on modified-live vaccine serotype 6 with exchanged outer shell proteins confer full protection in sheep against virulent BTV8. PLoS ONE 2012, 7, e44619. [Google Scholar] [CrossRef] [PubMed]
- Eschbaumer, M.; Wackerlin, R.; Rudolf, M.; Keller, M.; Konig, P.; Zemke, J.; Hoffmann, B.; Beer, M. Infectious blood or culture-grown virus: A comparison of bluetongue virus challenge models. Vet. Microbiol. 2010, 146, 150–154. [Google Scholar] [CrossRef]
- Bonneau, K.R.; DeMaula, C.D.; Mullens, B.A.; MacLachlan, N.J. Duration of viraemia infectious to culicoides sonorensis in bluetongue virus-infected cattle and sheep. Vet. Microbiol. 2002, 88, 115–125. [Google Scholar] [CrossRef]
- MacLachlan, N.J.; Crafford, J.E.; Vernau, W.; Gardner, I.A.; Goddard, A.; Guthrie, A.J.; Venter, E.H. Experimental reproduction of severe bluetongue in sheep. Vet. Pathol. 2008, 45, 310–315. [Google Scholar] [CrossRef]
- Gould, A.R.; Eaton, B.T. The amino acid sequence of the outer coat protein VP2 of neutralizing monoclonal antibody-resistant, virulent and attenuated bluetongue viruses. Virus Res. 1990, 17, 161–172. [Google Scholar] [CrossRef]
- Janowicz, A.; Caporale, M.; Shaw, A.; Gulletta, S.; Di Gialleonardo, L.; Ratinier, M.; Palmarini, M. Multiple genome segments determine virulence of bluetongue virus serotype 8. J. Virol. 2015, 89, 5238–5249. [Google Scholar] [CrossRef]
- Zhang, X.; Boyce, M.; Bhattacharya, B.; Zhang, X.; Schein, S.; Roy, P.; Zhou, Z.H. Bluetongue virus coat protein vp2 contains sialic acid-binding domains, and vp5 resembles enveloped virus fusion proteins. PNAS 2010, 107, 6292–6297. [Google Scholar] [CrossRef]
- Zhang, X.; Patel, A.; Celma, C.C.; Yu, X.; Roy, P.; Zhou, Z.H. Atomic model of a nonenveloped virus reveals pH sensors for a coordinated process of cell entry. Nat. Struct. Mol. Biol. 2016, 23, 74–80. [Google Scholar] [CrossRef]
- Tan, B.H.; Nason, E.; Staeuber, N.; Jiang, W.; Monastryrskaya, K.; Roy, P. Rgd tripeptide of bluetongue virus vp7 protein is responsible for core attachment to culicoides cells. J. Virol. 2001, 75, 3937–3947. [Google Scholar] [CrossRef] [PubMed]
- Kerviel, A.; Ge, P.; Lai, M.; Jih, J.; Boyce, M.; Zhang, X.; Zhou, Z.H.; Roy, P. Atomic structure of the translation regulatory protein ns1 of bluetongue virus. Nat. Microbiol. 2019, 4, 837–845. [Google Scholar] [CrossRef]
- Lymperopoulos, K.; Wirblich, C.; Brierley, I.; Roy, P. Sequence specificity in the interaction of bluetongue virus non-structural protein 2 (ns2) with viral RNA. J. Biol. Chem. 2003, 278, 31722–31730. [Google Scholar] [CrossRef] [PubMed]
- Mumtsidu, E.; Makhov, A.M.; Roessle, M.; Bathke, A.; Tucker, P.A. Structural features of the bluetongue virus ns2 protein. J. Struct. Biol. 2007, 160, 157–167. [Google Scholar] [CrossRef]
- Ratinier, M.; Shaw, A.E.; Barry, G.; Gu, Q.; Di Gialleonardo, L.; Janowicz, A.; Varela, M.; Randall, R.E.; Caporale, M.; Palmarini, M. Bluetongue virus ns4 protein is an interferon antagonist and a determinant of virus virulence. J. Virol. 2016, 90, 5427–5439. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, E.; Doceul, V.; Lara, E.; Breard, E.; Sailleau, C.; Vidalain, P.O.; Meurs, E.F.; Dabo, S.; Schwartz-Cornil, I.; Zientara, S.; et al. Ns3 of bluetongue virus interferes with the induction of type I interferon. J. Virol. 2013, 87, 8241–8246. [Google Scholar] [CrossRef]
- Beaton, A.R.; Rodriguez, J.; Reddy, Y.K.; Roy, P. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein ns3 and mediates virus release. PNAS 2002, 99, 13154–13159. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Harty, R.N. The ns3 protein of bluetongue virus exhibits viroporin-like properties. J. Biol. Chem. 2004, 279, 43092–43097. [Google Scholar] [CrossRef]
- Gambles, R.M. Bluetongue of sheep in Cyprus. J. Comparat. Pathol. Therap. 1949, 59, 176–190. [Google Scholar] [CrossRef]
- Nomikou, K.; Hughes, J.; Wash, R.; Kellam, P.; Breard, E.; Zientara, S.; Palmarini, M.; Biek, R.; Mertens, P. Widespread reassortment shapes the evolution and epidemiology of bluetongue virus following european invasion. PLoS Pathog. 2015, 11, e1005056. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, S.J.; McHolland, L.E.; Tabachnick, W.J. Cell lines from culicoides variipennis (diptera: Ceratopogonidae) support replication of bluetongue virus. J. Invertebr. Pathol. 1989, 54, 385–393. [Google Scholar] [CrossRef]
- Hooper, P.T.; Lunt, R.A.; Stanislawek, W.L. A trial comparing the virulence of some South African and australian bluetongue viruses. Aust. Vet. J. 1996, 73, 36–37. [Google Scholar] [CrossRef] [PubMed]
- Karber, G. Beitrag zur kollektiven behandlung pharmakologischer reihenversuche. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Clavijo, A.; Heckert, R.A.; Dulac, G.C.; Afshar, A. Isolation and identification of bluetongue virus. J. Virol. Methods 2000, 87, 13–23. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Perales, C.; Rodriguez-Frias, F.; Esteban, J.I.; Quer, J.; Domingo, E. Viral quasispecies complexity measures. Virology 2016, 493, 227–237. [Google Scholar] [CrossRef]
- Makanya, A.N.; Dimova, I.; Koller, T.; Styp-Rekowska, B.; Djonov, V. Dynamics of the developing chick chorioallantoic membrane assessed by stereology, allometry, immunohistochemistry and molecular analysis. PLoS ONE 2016, 11, e0152821. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.H.; Coles, J.D.W.A.; Alexander, R.A. Cultivation of bluetongue virus in fertile eggs produced on a vitamin-deficient diet. Nature 1940, 145, 1022. [Google Scholar] [CrossRef]
- White, P.T. Experimental studies on the circulatory system of the late chick embryo. J. Exp. Biol. 1974, 61, 571–592. [Google Scholar] [PubMed]
- Nowak-Sliwinska, P.; Segura, T.; Iruela-Arispe, M.L. The chicken chorioallantoic membrane model in biology, medicine and bioengineering. Angiogenesis 2014, 17, 779–804. [Google Scholar] [CrossRef]
- Karpala, A.J.; Bagnaud-Baule, A.; Goossens, K.E.; Lowenthal, J.W.; Bean, A.G. Ontogeny of the interferon system in chickens. J. Reprod. Immunol. 2012, 94, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Darpel, K.E.; Monaghan, P.; Simpson, J.; Anthony, S.J.; Veronesi, E.; Brooks, H.W.; Elliott, H.; Brownlie, J.; Takamatsu, H.H.; Mellor, P.S.; et al. Involvement of the skin during bluetongue virus infection and replication in the ruminant host. Vet. Res. 2012, 43, 40. [Google Scholar] [CrossRef]
- Majesky, M.W. Vascular development. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e17–e24. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, A.S.; Stott, J.L.; Squire, K.R.; Osburn, B.I. Strain-dependent virulence characteristics of bluetongue virus serotype 11. J. Gen. Virol. 1986, 67, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.A.; de Mattos, C.C.; de Mattos, C.A.; Osburn, B.I. Association of bluetongue virus gene segment 5 with neuroinvasiveness. J. Virol. 1994, 68, 1255–1257. [Google Scholar]
- Hooper, P.T.; Lunt, R.A.; Gould, A.R.; Hyatt, A.D.; Russell, G.M.; Kattenbelt, J.A.; Blacksell, S.D.; Reddacliff, L.A.; Kirkland, P.D.; Davis, R.J.; et al. Epidemic of blindness in kangaroos - evidence of a viral aetiology. Aust. Vet. J. 1999, 77, 529–536. [Google Scholar] [CrossRef]
- Attoui, H.; Mendez-Lopez, M.R.; Rao, S.; Hurtado-Alendes, A.; Lizaraso-Caparo, F.; Jaafar, F.M.; Samuel, A.R.; Belhouchet, M.; Pritchard, L.I.; Melville, L.; et al. Peruvian horse sickness virus and yunnan orbivirus, isolated from vertebrates and mosquitoes in Peru and Australia. Virology 2009, 394, 298–310. [Google Scholar] [CrossRef]
- Erasmus, B.J. Preliminary observations of the value of the guinea-pig in determining the innocuity and antigenicity of neurotropic attenuated horsesickness strains. Onderstepoort. J. Vet. Res. 1963, 30, 11–22. [Google Scholar]
- Taylor, M.B.; van der Meyden, C.H.; Erasmus, B.J.; Reid, R.; Labuscagne, J.H.; Dreyer, L.; Prozesky, O.W. Encephalitis and chorioretinitis associated with neurotropic african horsesickness virus infection in laboratory workers. Part IV. Experimental infection of the vervet monkey (cercopithecus pygerythrus). S Afr. Med. J. 1992, 81, 462–467. [Google Scholar]
- Feenstra, F.; van Rijn, P.A. Current and next-generation bluetongue vaccines: Requirements, strategies, and prospects for different field situations. Crit. Rev. Microbiol. 2017, 43, 142–155. [Google Scholar] [CrossRef]
- Coffey, L.L.; Vasilakis, N.; Brault, A.C.; Powers, A.M.; Tripet, F.; Weaver, S.C. Arbovirus evolution in vivo is constrained by host alternation. PNAS 2008, 105, 6970–6975. [Google Scholar] [CrossRef]
- Bernard, K.A.; Klimstra, W.B.; Johnston, R.E. Mutations in the e2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 2000, 276, 93–103. [Google Scholar] [CrossRef]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. PNAS 1999, 96, 13910–13913. [Google Scholar] [CrossRef]
- Brackney, D.E.; Schirtzinger, E.E.; Harrison, T.D.; Ebel, G.D.; Hanley, K.A. Modulation of flavivirus population diversity by RNA interference. J. Virol. 2015, 89, 4035–4039. [Google Scholar] [CrossRef]
- Schnettler, E.; Ratinier, M.; Watson, M.; Shaw, A.E.; McFarlane, M.; Varela, M.; Elliott, R.M.; Palmarini, M.; Kohl, A. RNA interference targets arbovirus replication in culicoides cells. J. Virol. 2013, 87, 2441–2454. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Vignuzzi, M. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 2011, 85, 1025–1035. [Google Scholar] [CrossRef]
- Drolet, B.S.; Reister, L.M.; Rigg, T.D.; Nol, P.; Podell, B.K.; Mecham, J.O.; VerCauteren, K.C.; van Rijn, P.A.; Wilson, W.C.; Bowen, R.A. Experimental infection of white-tailed deer (odocoileus virginianus) with northern european bluetongue virus serotype 8. Vet. Microbiol. 2013, 166, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Maan, S.; Maan, N.S.; Ross-smith, N.; Batten, C.A.; Shaw, A.E.; Anthony, S.J.; Samuel, A.R.; Darpel, K.E.; Veronesi, E.; Oura, C.A.; et al. Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains. Virology 2008, 377, 308–318. [Google Scholar] [CrossRef]
- Caporale, M.; Wash, R.; Pini, A.; Savini, G.; Franchi, P.; Golder, M.; Patterson-Kane, J.; Mertens, P.; Di Gialleonardo, L.; Armillotta, G.; et al. Determinants of bluetongue virus virulence in murine models of disease. J. Virol. 2011, 85, 11479–11489. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment (Protein) | Sh | Kc | Kb2 | Kb3 | Eab2 | Eab3 |
|---|---|---|---|---|---|---|
| 2 (VP2) | - | - | - | - | G2501A (G827E) | G2501A (G827E) |
| 5 (NS1) | - | - | - | - | A315G (E94G) | A315G (E94G) |
| 6 (VP5) | - | - | C585T (T186I) | C585T (T186I) | A718G | A718G |
| 7 (VP7) | - | - | - | - | - | - |
| 8 (NS2) | - | A528G (Q169R) | A528G (Q169R) | A528G (Q169R) | A528G (Q169R) | |
| 9 (VP6) | - | - | A30G (M5I) | A30G (M5I) | A30G (M5I) | A30G (M5I) |
| C113T (T33M) | C113T (T33M) | |||||
| G189A | G189A | |||||
| 9 (NS4) | - | - | G189A (R3K) | G189A (R3K) | - | - |
| 10 (NS3/3) | - | - | - | - | - | - |
| Sh | Kc | Kb2 | Kb3 | Eab2 | Eab3 | |
|---|---|---|---|---|---|---|
| Seg-2 | 52.9 * | 58.6 | 43.7 | 50.7 | 60.0 | 66.3 |
| Seg-5 | 40.8 | 47.7 | 38.5 | 43.3 | 44.8 | 50.4 |
| Seg-6 | 55.2 | 57.1 | 41.7 | 46.5 | 55.5 | 58.7 |
| Seg-7 | 68.6 | 66.8 | 63.5 | 61.4 | 62.7 | 60.7 |
| Seg-8 | 40.7 | 47.1 | 43.1 | 45.1 | 44.3 | 51.0 |
| Seg-9 | 37.9 | 49.6 | 39.0 | 43.4 | 43.3 | 45.0 |
| Seg-10 | 50.1 | 40.8 | 37.1 | 40.8 | 43.3 | 49.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lean, F.Z.X.; Neave, M.J.; White, J.R.; Payne, J.; Eastwood, T.; Bergfeld, J.; Di Rubbo, A.; Stevens, V.; Davies, K.R.; Devlin, J.; et al. Attenuation of Bluetongue Virus (BTV) in an in ovo Model Is Related to the Changes of Viral Genetic Diversity of Cell-Culture Passaged BTV. Viruses 2019, 11, 481. https://doi.org/10.3390/v11050481
Lean FZX, Neave MJ, White JR, Payne J, Eastwood T, Bergfeld J, Di Rubbo A, Stevens V, Davies KR, Devlin J, et al. Attenuation of Bluetongue Virus (BTV) in an in ovo Model Is Related to the Changes of Viral Genetic Diversity of Cell-Culture Passaged BTV. Viruses. 2019; 11(5):481. https://doi.org/10.3390/v11050481
Chicago/Turabian StyleLean, Fabian Z. X., Matthew J. Neave, John R. White, Jean Payne, Teresa Eastwood, Jemma Bergfeld, Antonio Di Rubbo, Vittoria Stevens, Kelly R. Davies, Joanne Devlin, and et al. 2019. "Attenuation of Bluetongue Virus (BTV) in an in ovo Model Is Related to the Changes of Viral Genetic Diversity of Cell-Culture Passaged BTV" Viruses 11, no. 5: 481. https://doi.org/10.3390/v11050481
APA StyleLean, F. Z. X., Neave, M. J., White, J. R., Payne, J., Eastwood, T., Bergfeld, J., Di Rubbo, A., Stevens, V., Davies, K. R., Devlin, J., Williams, D. T., & Bingham, J. (2019). Attenuation of Bluetongue Virus (BTV) in an in ovo Model Is Related to the Changes of Viral Genetic Diversity of Cell-Culture Passaged BTV. Viruses, 11(5), 481. https://doi.org/10.3390/v11050481