Phylogenetic Characterization of the Palyam Serogroup Orbiviruses



Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Nucleotide Sequences
3.2. Amino Acid Sequences
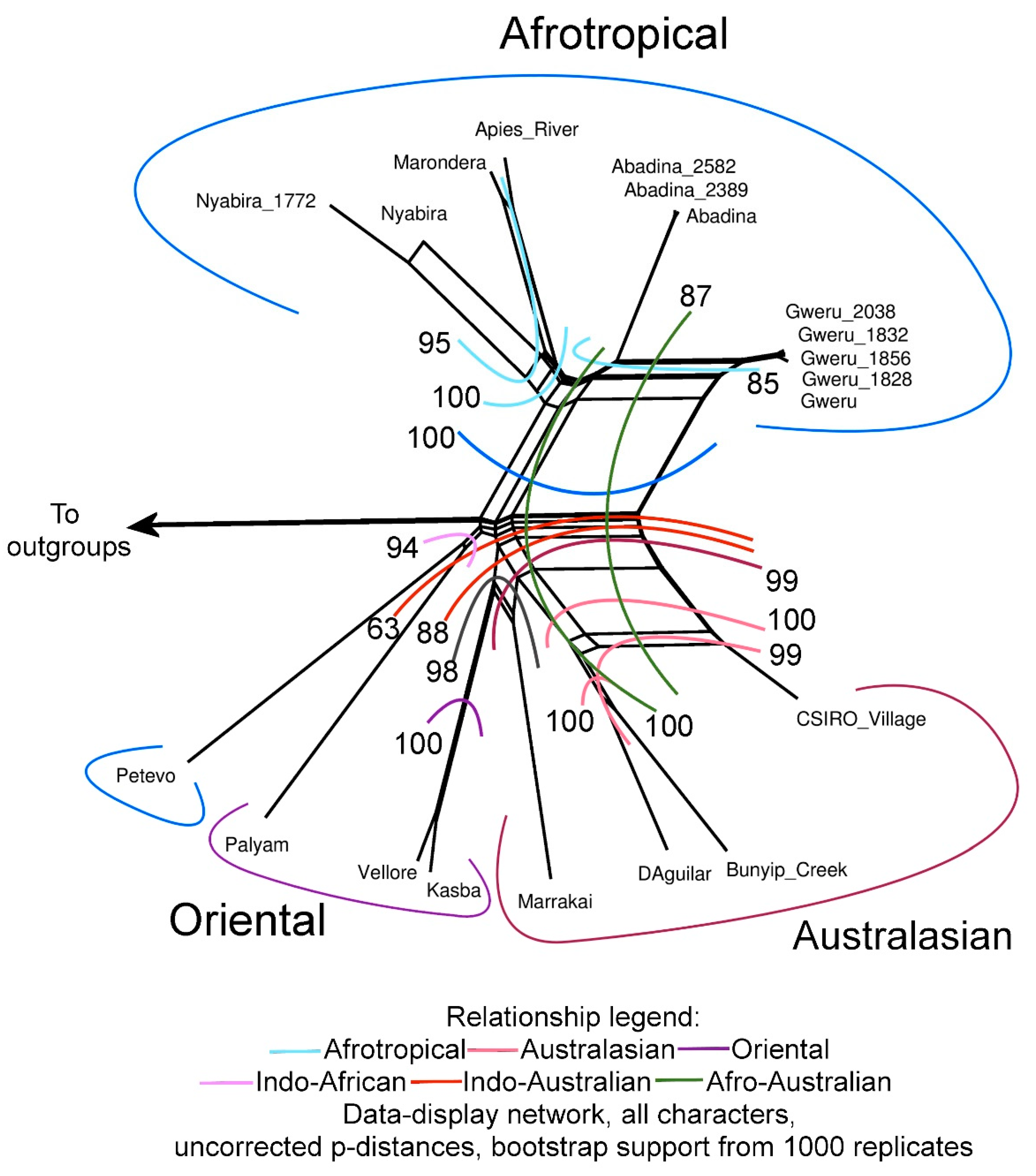
3.3. Phylogenetic Trees
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Aradaib, I.E.; Mohamed, M.E.H.; Abdalla, M.A. A single-tube RT-PCR for rapid detection and differentiation of some African isolates of Palyam serogroup orbiviruses. J. Virol. Methods 2009, 161, 70–74. [Google Scholar] [CrossRef]
- Swanepoel, R. Palyam serogroup orbivirus infections. In Infectious Diseases of Livestock, 2nd ed.; Coetzer, J.A.W., Tustin, R.C., Eds.; Oxford University Press: Cape Town, South Africa, 2004; pp. 1252–1255. [Google Scholar]
- Yamakawa, M.; Kubo, M.; Furuuchi, S. Molecular analysis of the genome of Chuzan virus, a member of the Palyam serogroup viruses, and its phylogenetic relationships to other orbiviruses. J. Gen. Virol. 1999, 80, 937–941. [Google Scholar] [CrossRef][Green Version]
- Miura, Y.; Kubo, M.; Goto, Y.; Kono, Y. Chuzan disease as congenital-hydranencephaly cerebellar hypoplasia syndrome in calves. Jpn. Agric. Res. Q. 1991, 25, 55–60. [Google Scholar]
- St George, T.D. An overview of arboviruses affecting domestic animals in Australia. Aust Vet. J. 1989, 66, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Miura, Y.; Kono, Y. Epidemiological survey of an epidemic of congenital abnormalities with hydranencephaly-cerebellar hypoplasia syndrome of calves occurring in 1985/86 and seroepidemiological investigations on Chuzan virus, a putative causal agent of the disease, in Japan. Jpn. J. Vet. Sci. 1988, 50, 405–413. [Google Scholar] [CrossRef][Green Version]
- Whistler, T.; Swanepoel, R. Characterization of potentially foetotropic Palyam serogroup orbiviruses isolated in Zimbabwe. J. Gen. Virol. 1988, 69, 2221–2227. [Google Scholar] [CrossRef]
- Whistler, T.; Swanepoel, R. Proteins of Palyam serogroup viruses. J. Gen. Virol. 1990, 71, 1333–1338. [Google Scholar] [CrossRef]
- Yamakawa, M.; Furuuchi, S. Expression and antigenic characterization of the major core protein VP7 of Chuzan virus, a member of the Palyam serogroup orbiviruses. Vet. Microbiol. 2001, 83, 333–341. [Google Scholar] [CrossRef]
- Yamakawa, M.; Furuuchi, S.; Minobe, Y. Molecular characterization of double-stranded RNA segments encoding the major capsid proteins of a Palyam serogroup orbivirus that caused an epizootic of congenital abnormalities in cattle. J. Gen. Virol. 1999, 80, 205–208. [Google Scholar] [CrossRef]
- Harasawa, R.; Yoshida, T.; Iwashita, O.; Goto, Y.; Miura, Y. Biochemical characteristics of Chuzan virus, a new serotype of Palyam serogroup. Jpn. J. Vet. Sci. 1988, 50, 777–782. [Google Scholar] [CrossRef]
- Belhouchet, M.; Mohd Jaafar, F.; Firth, A.E.; Grimes, J.M.; Mertens, P.C.C.; Attoui, H. Detection of a fourth orbivirus non-structural protein. PLoS ONE 2011, 6, e25697. [Google Scholar] [CrossRef]
- Zwart, L.; Potgieter, C.A.; Clift, S.J.; van Staden, V. Characterising non-structural protein NS4 of African horse sickness virus. PLoS ONE 2015, 10, e0124281. [Google Scholar] [CrossRef]
- Van Staden, V.; Huismans, H. A comparison of the genes which encode non-structural NS3 of different orbiviruses. J. Gen. Virol. 1991, 72, 1073–1079. [Google Scholar] [CrossRef]
- Wang, F.; Lin, J.; Chang, J.; Cao, Y.; Qin, S.; Wu, J.; Yu, L. Isolation, complete genome sequencing, and phylogenetic analysis of the first Chuzan virus in China. Virus Genes 2016, 52, 138–141. [Google Scholar] [CrossRef]
- Yang, H.; Xiao, L.; Meng, J.; Xiong, H.; Gao, L.; Liao, D.; Li, H. Complete genome sequence of a Chuzan virus strain isolated for the first time in mainland China. Arch. Virol. 2016, 161, 1073–1077. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ohashi, S.; Kanno, T.; Yamazoe, R.; Yoshida, K.; Tsuda, T.; Sakamoto, K. Genetic diversity of RNA segments 5, 7 and 9 of the Palyam serogroup orbiviruses from Japan, Australia and Zimbabwe. Virus Res. 2000, 68, 145–153. [Google Scholar] [CrossRef]
- Buaro, I.; Lomonosov, N.N.; Votiakov, V.I.; Samoĭlova, T.I.; Poleshchuk, N.N. Kindia virus—A new arbovirus isolated in the territory of the Republic of Guinea. Vopr. Virusol. 1987, 32, 109–112. [Google Scholar]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; Dijk, A.A.V. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef]
- Maan, S.; Rao, S.; Maan, N.S.; Anthony, S.J.; Attoui, H.; Samual, A.R.; Mertens, P.P.C. Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J. Virol. Methods 2007, 143, 132–139. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Swartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop, New Orleans, LA, USA, 4 November 2010; pp. 1–8. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30(9), 1312–1313. [Google Scholar] [CrossRef]
- Ohashi, S.; Matsumori, Y.; Yanase, T.; Yamakawa, M.; Kato, T.; Tsuda, T. Evidence of an antigenic shift among Palyam serogroup orbiviruses. J. Clin. Microbiol. 2004, 42, 4610–4614. [Google Scholar] [CrossRef]
- Maan, S.; Maan, N.S.; Nomikou, K.; Veronesi, E.; Bachanek-Bankowska, K.; Belaganahalli, M.N.; Attoui, H.; Mertens, P.P.C. Complete genome characterization of a novel 26th bluetongue virus serotype from Kuwait. PLoS ONE 2011, 6, e26147. [Google Scholar] [CrossRef]
- Maan, S.; Maan, N.S.; van Rijn, P.A.; van Gennip, R.G.P.; Sanders, A.; Wright, I.M. Full genome characterization of bluetongue virus serotype 6 from the Netherlands 2008 and comparison to other field and vaccine strains. PLoS ONE 2010, 5, e10323. [Google Scholar] [CrossRef]
- Whistler, T.; Swanepoel, R.; Erasmus, B.J. Characterization of Palyam serogroup orbiviruses isolated in South Africa and serologic evidence for their widespread distribution in the country. Epidemiol. Infect. 1989, 102, 317–324. [Google Scholar] [CrossRef]
- Bodkin, D.K.; Knudson, D.L. Genetic relatedness of Palyam serogroup viruses by RNA-RNA blot hybridization. J. Gen. Virol. 1986, 67, 683–691. [Google Scholar] [CrossRef]
- Saluzzo, J.F.; Digoutte, J.P.; Cornet, J.P.; Heme, C.; Herve, J.P.; Gonzalez, J.P.; Georges, A.J. Petevo virus, a new arbovirus of the Palyam group isolated in Central African Republic from the tick Amblyomma variegatum. Ann. Virol. 1982, 133, 215–222. [Google Scholar]
- Eagles, D.; Deveson, T.; Walker, P.J.; Zalucki, M.P.; Durr, P. Evaluation of long-distance dispersal of Culicoides midges into northern Australia using a migration model. Med. Vet. Entomol. 2012, 26, 334–340. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Serotype | Strain Designation | Country of Isolation | Source of Isolation | Year of Initial Isolation |
|---|---|---|---|---|
| Prototypes | ||||
| Abadina | Ib Ar 22388 | Nigeria | Culicoides spp. | 1967 |
| Bunyip Creek | CSIRO 87 | Australia | Culicoides schultzei | 1976 |
| CSIRO Village | CSIRO 11 | Australia | Culicoides spp. | 1974 |
| D’Aguilar | B8112 | Australia | Culex brevitarsus | 1968 |
| Kasba | GG15534 | India | Culex vishnui | 1957 |
| Marrakai | CSIRO 82 | Australia | Culicoides spp. | 1975 |
| Palyam | G5287 | India | Culex vishnui | 1956 |
| Petevo | Ar TB 2032 | Central African Republic | Amblyomma variegatum | 1978 |
| Vellore | 68886 | India | Culex pseudovishnui | 1956 |
| Apies River | O/4518 | South Africa | Cow blood | 2005 |
| Gweru | VRL1726/76 | Zimbabwe | Bovine foetus | 1976 |
| Marondera | VRL1070/78 | Zimbabwe | Cow viscera | 1978 |
| Nyabira | VRL792/73 | Zimbabwe | Bovine foetus | 1973 |
| Field isolates | ||||
| Abadina 2582 | 2582/78 | Zimbabwe | Bovine foetus | 1978 |
| Abadina 2389 | 2389/76 | Zimbabwe | Bovine foetus | 1976 |
| Nyabira 1772 | 1772/74 | Zimbabwe | Bovine foetus | 1974 |
| Gweru 1828 | 1828/82 | Zimbabwe | Bovine foetus | 1982 |
| Gweru 1832 | 1832/79 | Zimbabwe | Bovine foetus | 1979 |
| Gweru 2038 | 2038/76 | Zimbabwe | Bovine foetus | 1976 |
| Gweru 1856 | 1856/76 | Zimbabwe | Bovine foetus | 1976 |
| Virus isolate | Segment 1 (VP1) | Segment 2 (VP2) | Segment 3 (VP3) | Segment 4 (VP4) | Segment6 (VP5) | Segment 9 (VP6) | Segment 7 (VP7) | Segment5 (NS1) | Segment8 (NS2) | Segment 10 (NS3) |
|---|---|---|---|---|---|---|---|---|---|---|
| Abadina | MH782454 | MH823477 | MH817078 | MH817097 | MH817117 | MH823377 | MH823397 | MH823417 | MH823437 | MH823457 |
| Bunyip Creek | MH782455 | MH823478 | MH817079 | MH817098 | MH817118 | MH823378 | MH823398 | MH823418 | MH823438 | MH823458 |
| CSIRO Village | MH782456 | MH823479 | MH817080 | MH817099 | MH817119 | MH823379 | MH823399 | MH823419 | MH823439 | MH823459 |
| D’Aguilar | MH782457 | MH823480 | MH817081 | MH817100 | MH817120 | MH823380 | MH823400 | MH823420 | MH823440 | MH823460 |
| Kasba | MH782458 | MH823481 | MH817082 | MH817101 | MH817121 | MH823381 | MH823401 | MH823421 | MH823441 | MH823461 |
| Marrakai | MH782459 | MH823482 | MH817083 | MH817102 | MH817122 | MH823382 | MH823402 | MH823422 | MH823442 | MH823462 |
| Palyam | MH782460 | MH823483 | MH817084 | MH817103 | MH817123 | MH823383 | MH823403 | MH823423 | MH823443 | MH823463 |
| Petevo | MH782461 | MH823484 | MH817085 | MH817104 | MH817124 | MH823384 | MH823404 | MH823424 | MH823444 | MH823464 |
| Vellore | MH782462 | MH823485 | MH817086 | MH817105 | MH817125 | MH823385 | MH823405 | MH823425 | MH823445 | MH823465 |
| Apies River | MH782463 | MH823486 | MH817087 | MH817106 | MH817126 | MH823386 | MH823406 | MH823426 | MH823446 | MH823466 |
| Gweru | MH782464 | MH823487 | MH817088 | MH817107 | MH817127 | MH823387 | MH823407 | MH823427 | MH823447 | MH823467 |
| Marondera | MH782465 | MH823488 | MH817089 | MH817108 | MH817128 | MH823388 | MH823408 | MH823428 | MH823448 | MH823468 |
| Nyabira | MH782466 | MH823489 | MH817090 | MH817109 | MH817129 | MH823389 | MH823409 | MH823429 | MH823449 | MH823469 |
| Abadina 2582 | MH782467 | MH823490 | MH817091 | MH817110 | MH817130 | MH823390 | MH823410 | MH823430 | MH823450 | MH823470 |
| Abadina 2389 | MH782468 | MH823491 | MH817092 | MH817111 | MH817131 | MH823391 | MH823411 | MH823431 | MH823451 | MH823471 |
| Gweru 1828 | MH782469 | MH823492 | MH817093 | MH817112 | MH817132 | MH823392 | MH823412 | MH823432 | MH823452 | MH823472 |
| Gweru 1832 | MH782470 | MH823493 | MH817094 | MH817113 | MH817133 | MH823393 | MH823413 | MH823433 | MH823453 | MH823473 |
| Gweru 2038 | MH782471 | MH823494 | MH817095 | MH817114 | MH817134 | MH823394 | MH823414 | MH823434 | MH823454 | MH823474 |
| Gweru 1856 | MH782472 | MH823495 | MH817096 | MH817115 | MH817135 | MH823395 | MH823415 | MH823435 | MH823455 | MH823475 |
| Nyabira 1172 | NA | MH823496 | MK007563 | MH817116 | MH817136 | MH823396 | MH823416 | MH823436 | MH823456 | MH823476 |
| AHS 1 Isolate E160445 | KX987198 | KX987199 | KX987200 | KX987201 | KX987202 | KX987203 | KX987204 | KX987205 | KX987206 | KX987207 |
| BTV Isolate BTV10 IND2003k3 | KP339244 | KP339245 | KP339246 | KP339247 | KP339248 | KP339249 | KP339250 | KP339251 | KP339252 | KP339253 |
| EEV Isolate Kimron1 | AB811635 | AB811636 | AB811637 | AB811638 | AB811639 | AB811630 | AB811631 | AB811632 | AB811633 | AB811634 |
| EHDV Isolate CC 304-06 | HM641772 | HM641773 | HM641774 | HM641775 | HM641777 | HM641700 | HM641778 | HM641776 | HM641779 | HM641781 |
| Virus | Abadina | Bunyip Creek | CSIRO Village | D’Aguilar | Kasba | Marrakai | Palyam | Petevo | Vellore | Apies River | Gweru | Marondera | Nyabira |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Abadina | 98.87 | 98.87 | 98.97 | 99.72 | 99.72 | 98.87 | 99.44 | 98.59 | 100.00 | 99.72 | 100.00 | 100.00 | |
| Bunyip Creek | 39.26 | 100.00 | 100.00 | 98.87 | 98.87 | 98.87 | 99.15 | 97.75 | 98.87 | 98.59 | 98.87 | 98.87 | |
| CSIRO Village | 51.95 | 37.86 | 100.00 | 98.87 | 98.87 | 98.87 | 99.15 | 97.75 | 98.87 | 98.59 | 98.87 | 98.87 | |
| D’Aguilar | 38.40 | 47.95 | 36.70 | 98.87 | 98.87 | 98.87 | 99.15 | 97.75 | 98.87 | 98.59 | 98.87 | 98.97 | |
| Kasba | 86.68 | 40.33 | 52.25 | 39.96 | 100.00 | 99.15 | 99.72 | 98.87 | 99.72 | 99.44 | 99.72 | 99.72 | |
| Marrakai | 42.72 | 39.25 | 44.80 | 37.91 | 43.01 | 99.15 | 99.72 | 98.87 | 99.72 | 99.44 | 99.72 | 99.72 | |
| Palyam | 37.63 | 46.78 | 35.85 | 95.82 | 38.80 | 36.83 | 99.44 | 98.03 | 98.87 | 98.59 | 98.87 | 98.87 | |
| Petevo | 43.94 | 38.45 | 44.31 | 36.14 | 44.42 | 52.85 | 35.26 | 98.59 | 99.44 | 99.15 | 99.44 | 99.44 | |
| Vellore | 85.19 | 39.65 | 51.07 | 39.57 | 98.11 | 42.33 | 38.41 | 43.84 | 98.59 | 98.31 | 98.59 | 98.59 | |
| Apies River | 39.07 | 74.26 | 38.25 | 47.37 | 40.23 | 38.48 | 46.30 | 37.29 | 39.85 | 99.72 | 100.00 | 100.00 | |
| Gweru | 51.95 | 37.86 | 99.90 | 36.70 | 52.25 | 44.80 | 35.85 | 44.31 | 51.07 | 38.25 | 99.72 | 99.72 | |
| Marondera | 38.59 | 74.17 | 38.15 | 46.98 | 37.75 | 38.48 | 45.91 | 37.00 | 39.36 | 98.72 | 38.15 | 100.00 | |
| Nyabira | 38.30 | 48.14 | 37.38 | 95.99 | 39.86 | 37.91 | 92.23 | 36.24 | 39.38 | 47.56 | 37.38 | 47.08 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebersohn, K.; Coetzee, P.; Snyman, L.P.; Swanepoel, R.; Venter, E.H. Phylogenetic Characterization of the Palyam Serogroup Orbiviruses. Viruses 2019, 11, 446. https://doi.org/10.3390/v11050446
Ebersohn K, Coetzee P, Snyman LP, Swanepoel R, Venter EH. Phylogenetic Characterization of the Palyam Serogroup Orbiviruses. Viruses. 2019; 11(5):446. https://doi.org/10.3390/v11050446
Chicago/Turabian StyleEbersohn, Karen, Peter Coetzee, Louwrens P. Snyman, Robert Swanepoel, and Estelle H. Venter. 2019. "Phylogenetic Characterization of the Palyam Serogroup Orbiviruses" Viruses 11, no. 5: 446. https://doi.org/10.3390/v11050446
APA StyleEbersohn, K., Coetzee, P., Snyman, L. P., Swanepoel, R., & Venter, E. H. (2019). Phylogenetic Characterization of the Palyam Serogroup Orbiviruses. Viruses, 11(5), 446. https://doi.org/10.3390/v11050446