Interpreting Viral Deep Sequencing Data with GLUE

 , , , , , ,
, , , , , ,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
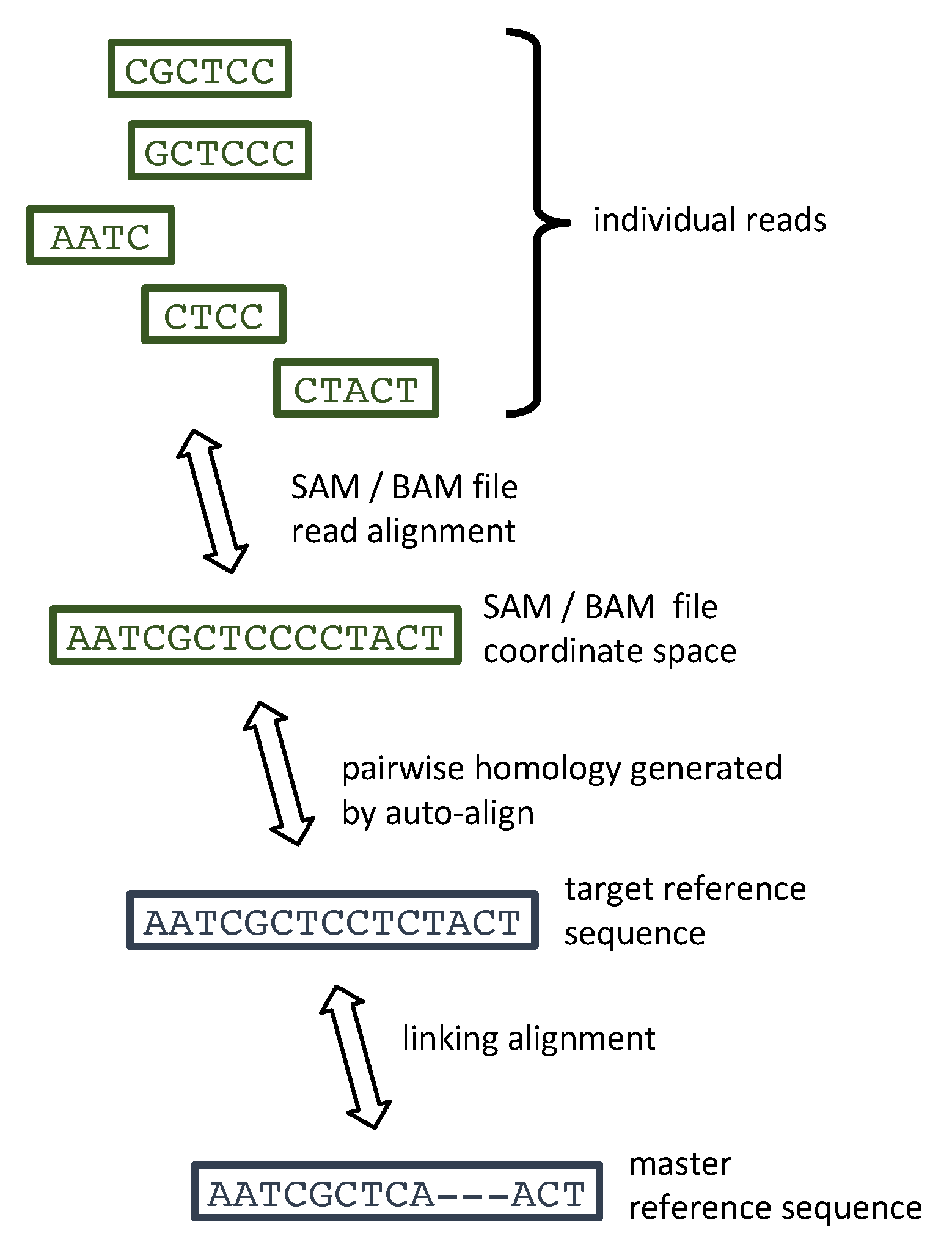
3. Materials and Methods
3.1. Sequencing Data
3.2. GLUE samReporter Design
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAM | Binary alignment mapping |
| DAA | Direct-acting Antiviral |
| DNA | Deoxyribonucleic Acid |
| ICTV | International Committee for the Taxonomy of Viruses |
| HCV | Hepatitis C virus |
| RAS | Resistance-associated Substitution |
| RNA | Ribonucleic Acid |
| SAM | Sequence alignment mapping |
| SNV | Single nucleotide variant |
| SVR | Sustained virological response |
References
- Holmes, E.C. The Evolution and Emergence of RNA Viruses; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Lee, W.P.; Stromberg, M.P.; Ward, A.; Stewart, C.; Garrison, E.P.; Marth, G.T. MOSAIK: A Hash-Based Algorithm for Accurate Next-Generation Sequencing Short-Read Mapping. PLoS ONE 2014, 9, e90581. [Google Scholar] [CrossRef]
- Lunter, G.; Goodson, M. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011, 21, 936–939. [Google Scholar] [CrossRef]
- Tanoti: A BLAST-Guided Reference-Based Short Read Aligner. Available online: http://www.bioinformatics.cvr.ac.uk/tanoti.php (accessed on 24 February 2019).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Yang, X.; Charlebois, P.; Gnerre, S.; Coole, M.G.; Lennon, N.J.; Levin, J.Z.; Qu, J.; Ryan, E.M.; Zody, M.C.; Henn, M.R. De novo assembly of highly diverse viral populations. BMC Genom. 2012, 13, 475. [Google Scholar] [CrossRef]
- Wymant, C.; Fraser, C.; Hall, M.; Golubchik, T.; Bannert, N.; Fellay, J.; Fransen, K.; Porter, K.; Gourlay, A.; Grabowski, M.K.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4, vey007. [Google Scholar] [CrossRef]
- Li, H.; Wysoker, A.; Handsaker, B.; Marth, G.; Abecasis, G.; Ruan, J.; Homer, N.; Durbin, R.; Fennell, T. The 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Macalalad, A.R.; Zody, M.C.; Charlebois, P.; Lennon, N.J.; Newman, R.M.; Malboeuf, C.M.; Ryan, E.M.; Boutwell, C.L.; Power, K.A.; Brackney, D.E.; et al. Highly Sensitive and Specific Detection of Rare Variants in Mixed Viral Populations from Massively Parallel Sequence Data. PLoS Comput. Biol. 2012, 8, e1002417. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Patrick, C.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant inference for viral populations. BMC Genom. 2013, 14, 674. [Google Scholar] [CrossRef]
- Sandmann, S.; de Graaf, A.O.; Karimi, M.; van der Reijden, B.A.; Hellström-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 43169. [Google Scholar] [CrossRef]
- Ibrahim, B.; McMahon, D.P.; Hufsky, F.; Beer, M.; Deng, L.; Mercier, P.L.; Palmarini, M.; Thiel, V.; Marz, M. A new era of virus bioinformatics. Virus Res. 2018, 251, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Combet, C.; Bukh, J.; Shin-I, T.; Deleage, G.; Mizokami, M.; Richardson, R.; Sablon, E.; Yusim, K.; Pawlotsky, J.M.; et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 2006, 44, 1355–1361. [Google Scholar] [CrossRef]
- Verbist, B.M.; Aerssens, J.; Reumers, J.; Thys, K.; Van der Borght, K.; Clement, L.; Thas, O.; Talloen, W.; Wetzels, Y. VirVarSeq: A low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2014, 31, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Döring, M.; Büch, J.; Friedrich, G.; Pironti, A.; Kalaghatgi, P.; Knops, E.; Heger, E.; Obermeier, M.; Däumer, M.; Thielen, A.; et al. geno2pheno[ngs-freq]: a genotypic interpretation system for identifying viral drug resistance using next-generation sequencing data. Nucleic Acids Res. 2018, 46, W271–W277. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, M.; Sloan, W.T.; Quince, C. Benchmarking of viral haplotype reconstruction programmes: An overview of the capacities and limitations of currently available programmes. Brief. Bioinform. 2012, 15, 431–442. [Google Scholar] [CrossRef]
- Singer, J.B.; Thomson, E.C.; McLauchlan, J.; Hughes, J.; Gifford, R.J. GLUE: A flexible software system for virus sequence data. BMC Bioinform. 2018, 19, 532. [Google Scholar] [CrossRef] [PubMed]
- Vermehren, J.; Park, J.S.; Jacobson, I.M.; Zeuzem, S. Challenges and perspectives of direct antivirals for the treatment of hepatitis C virus infection. J. Hepatol. 2018, 69, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Maio, V.C.D.; Howe, A.Y.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updates 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Recommendations on Treatment of Hepatitis C 2016. J. Hepatol. 2017, 66, 153–194. [Google Scholar] [CrossRef] [PubMed]
- IUPAC-IUB Commission on Biochemical Nomenclature (CBN). Abbreviations and symbols for nucleic acids, polynucleotides and their constituents. Recommendations 1970. Biochem. J. 1970, 120, 449–454. [Google Scholar] [CrossRef]
- HCV-GLUE: A Sequence Data Resource for Hepatitis C Virus. Available online: http://hcv.glue.cvr.ac.uk (accessed on 24 February 2019).
- Kalaghatgi, P.; Sikorski, A.M.; Knops, E.; Rupp, D.; Sierra, S.; Heger, E.; Neumann-Fraune, M.; Beggel, B.; Walker, A.; Timm, J.; et al. Geno2pheno[HCV]—A Web-based Interpretation System to Support Hepatitis C Treatment Decisions in the Era of Direct-Acting Antiviral Agents. PLoS ONE 2016, 11, e0155869. [Google Scholar] [CrossRef]
- Batty, E.M.; Wong, T.H.N.; Trebes, A.; Argoud, K.; Attar, M.; Buck, D.; Ip, C.L.C.; Golubchik, T.; Cule, M.; Bowden, R.; et al. A Modified RNA-Seq Approach for Whole Genome Sequencing of RNA Viruses from Faecal and Blood Samples. PLoS ONE 2013, 8, e66129. [Google Scholar] [CrossRef] [PubMed]
- Lamble, S.; Batty, E.; Attar, M.; Buck, D.; Bowden, R.; Lunter, G.; Crook, D.; El-Fahmawi, B.; Piazza, P. Improved workflows for high throughput library preparation using the transposome-based Nextera system. BMC Biotechnol. 2013, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Davalieva, K.; Kiprijanovska, S.; Plaseska-Karanfilska, D. Fast, reliable and low cost user-developed protocol for detection, quantification and genotyping of hepatitis C virus. J. Virol. Methods 2014, 196, 104–112. [Google Scholar] [CrossRef]
- Bonsall, D.; Ansari, M.; Ip, C.; Trebes, A.; Brown, A.; Klenerman, P.; Buck, D.; STOP-HCV Consortium; Piazza, P.; Barnes, E.; Bowden, R. ve-SEQ: Robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens [version 1; referees: 2 approved, 1 approved with reservations]. F1000Research 2015, 4, 1062. [Google Scholar] [CrossRef] [PubMed]
- TrimGalore: A Wrapper around Cutadapt and FastQC to Consistently Apply Adapter and Quality Trimming to FastQ Files, with Extra Functionality for RRBS Data. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 24 February 2019).
- Sreenu, V.B.; (MRC-University of Glasgow Centre for Virus Research, Glasgow, UK). Personal Communication, 2019.
- Gaidatzis, D.; Lerch, A.; Hahne, F.; Stadler, M.B. QuasR: Quantification and annotation of short reads in R. Bioinformatics 2014, 31, 1130–1132. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- V-FAT: A Post-Assembly Pipeline for the Finishing and Annotation of Viral Genomes. Available online: https://www.broadinstitute.org/viral-genomics/v-fat (accessed on 24 February 2019).
- SMALT: A Mapper for DNA Sequencing Reads. Available online: https://www.sanger.ac.uk/science/tools/smalt-0 (accessed on 24 February 2019).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Poordad, F.; Pol, S.; Asatryan, A.; Buti, M.; Shaw, D.; Hézode, C.; Felizarta, F.; Reindollar, R.W.; Gordon, S.C.; Pianko, S.; et al. Glecaprevir/Pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 2018, 67, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- A Java API for High-Throughput Sequencing Data (HTS) Formats. Available online: http://samtools.github.io/htsjdk/ (accessed on 24 February 2019).
- DiversiTools: Tool for Analysing Viral Diversity from HTS. Available online: http://josephhughes.github.io/DiversiTools/ (accessed on 24 February 2019).


{kind=link}
| Sequencing | Sample | Subtype | Virus | Codon | Ambiguous | Typical | Possible | Confirmed |
|---|---|---|---|---|---|---|---|---|
| Facility | ID | Protein | Location | Triplet | Residue (s) | Residues Set | Residues Set | |
| Glasgow | HCV294 | 3b | NS5B | 282 | WSY | S | CST | S |
| Glasgow | HCV300 | 3a | NS5A | 30 | RMG | A | AEKT | AK |
| PHE | R127 | 1a | NS5A | 24 | RSG | K | AGRT | GT |
| PHE | R164 | 3a | NS5A | 30 | RMG | A | AEKT | AK |
| PHE | R25 | 4r | NS5B | 159 | YTM | L | FL | L |
| PHE | R25 | 4r | NS5B | 282 | WSC | S | CST | S |
| PHE | R36 | 4r | NS5B | 282 | WSC | S | CST | S |
| PHE | R67 | 1a | NS5A | 30 | YAW | Q | HQY | QY |
| PHE | R91 | 1a | NS5A | 28 | RYG | M | AMTV | MV |
| Oxford | 7444 | 3a | NS5A | 62 | SYA | ST | ALPV | AL |
| Command | Description |
|---|---|
| nucleotide | Generate a table of nucleotide frequencies within a specific genome region. |
| depth | Generate a table of read depths within a specific genome region. |
| nucleotide-consensus | Generate a FASTA consensus file, optionally using ambiguity codes. |
| amino-acid | Generate a table of amino acid residue frequencies within a specific protein-coding region. |
| codon-triplets | Generate a table of codon frequencies within a specific protein-coding region. |
| variation scan | Scan for the presence or absence of GLUE Variations within reads. |
| export nucleotide-alignment | Export a specific part of the SAM alignment as a FASTA file. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singer, J.B.; Thomson, E.C.; Hughes, J.; Aranday-Cortes, E.; McLauchlan, J.; da Silva Filipe, A.; Tong, L.; Manso, C.F.; Gifford, R.J.; Robertson, D.L.; et al. Interpreting Viral Deep Sequencing Data with GLUE. Viruses 2019, 11, 323. https://doi.org/10.3390/v11040323
Singer JB, Thomson EC, Hughes J, Aranday-Cortes E, McLauchlan J, da Silva Filipe A, Tong L, Manso CF, Gifford RJ, Robertson DL, et al. Interpreting Viral Deep Sequencing Data with GLUE. Viruses. 2019; 11(4):323. https://doi.org/10.3390/v11040323
Chicago/Turabian StyleSinger, Joshua B., Emma C. Thomson, Joseph Hughes, Elihu Aranday-Cortes, John McLauchlan, Ana da Silva Filipe, Lily Tong, Carmen F. Manso, Robert J. Gifford, David L. Robertson, and et al. 2019. "Interpreting Viral Deep Sequencing Data with GLUE" Viruses 11, no. 4: 323. https://doi.org/10.3390/v11040323
APA StyleSinger, J. B., Thomson, E. C., Hughes, J., Aranday-Cortes, E., McLauchlan, J., da Silva Filipe, A., Tong, L., Manso, C. F., Gifford, R. J., Robertson, D. L., Barnes, E., Ansari, M. A., Mbisa, J. L., Bibby, D. F., Bradshaw, D., & Smith, D. (2019). Interpreting Viral Deep Sequencing Data with GLUE. Viruses, 11(4), 323. https://doi.org/10.3390/v11040323