Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses



Abstract
1. Introduction
Conserved RNA Structures Mediate Pathogenesis
2. Materials and Methods
2.1. Phylogeny Reconstruction
2.2. Structural Homology Search with Covariance Models
2.3. De novo Discovery of Conserved RNA Elements
3. Results
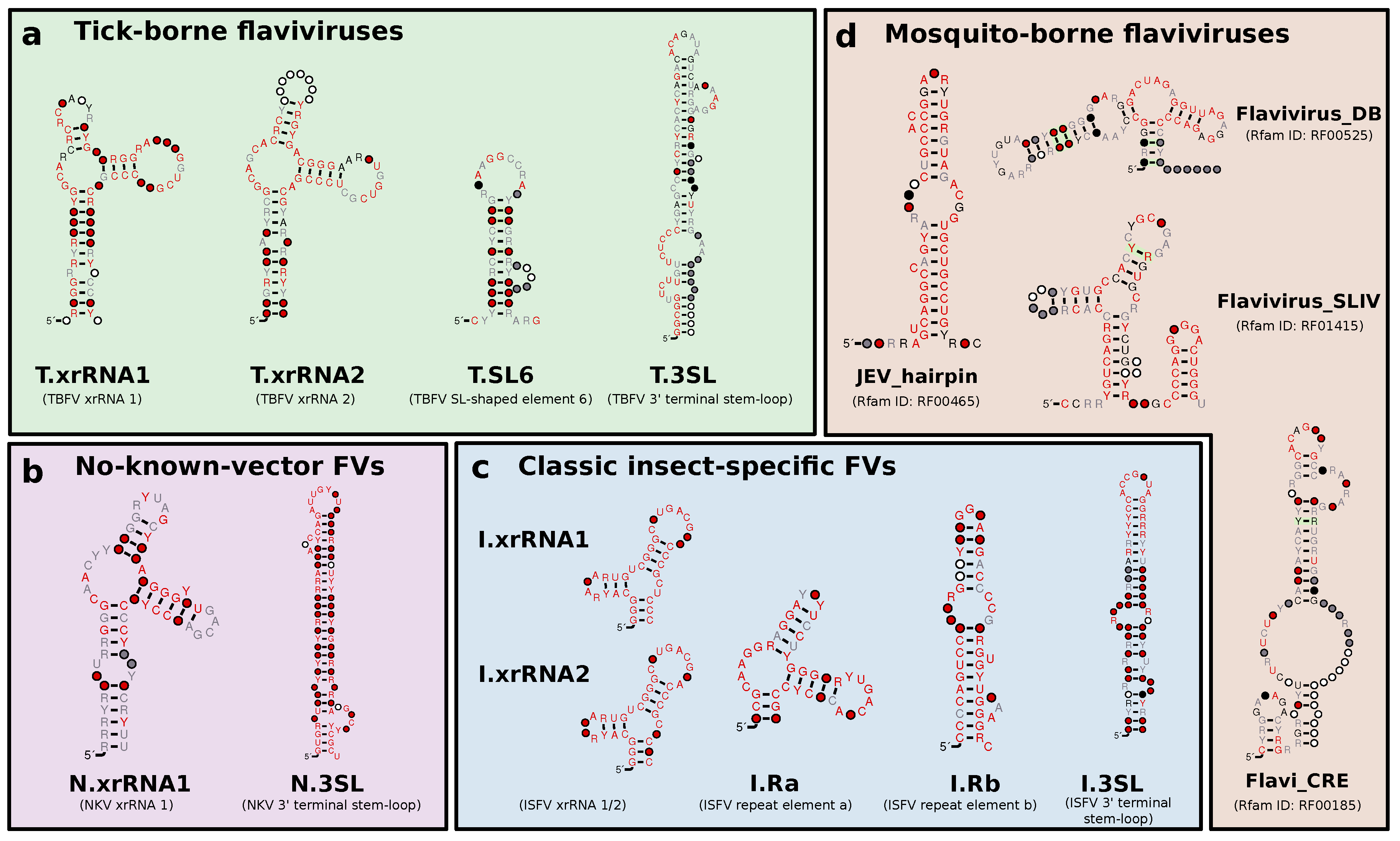
3.1. Construction of Seed Alignments
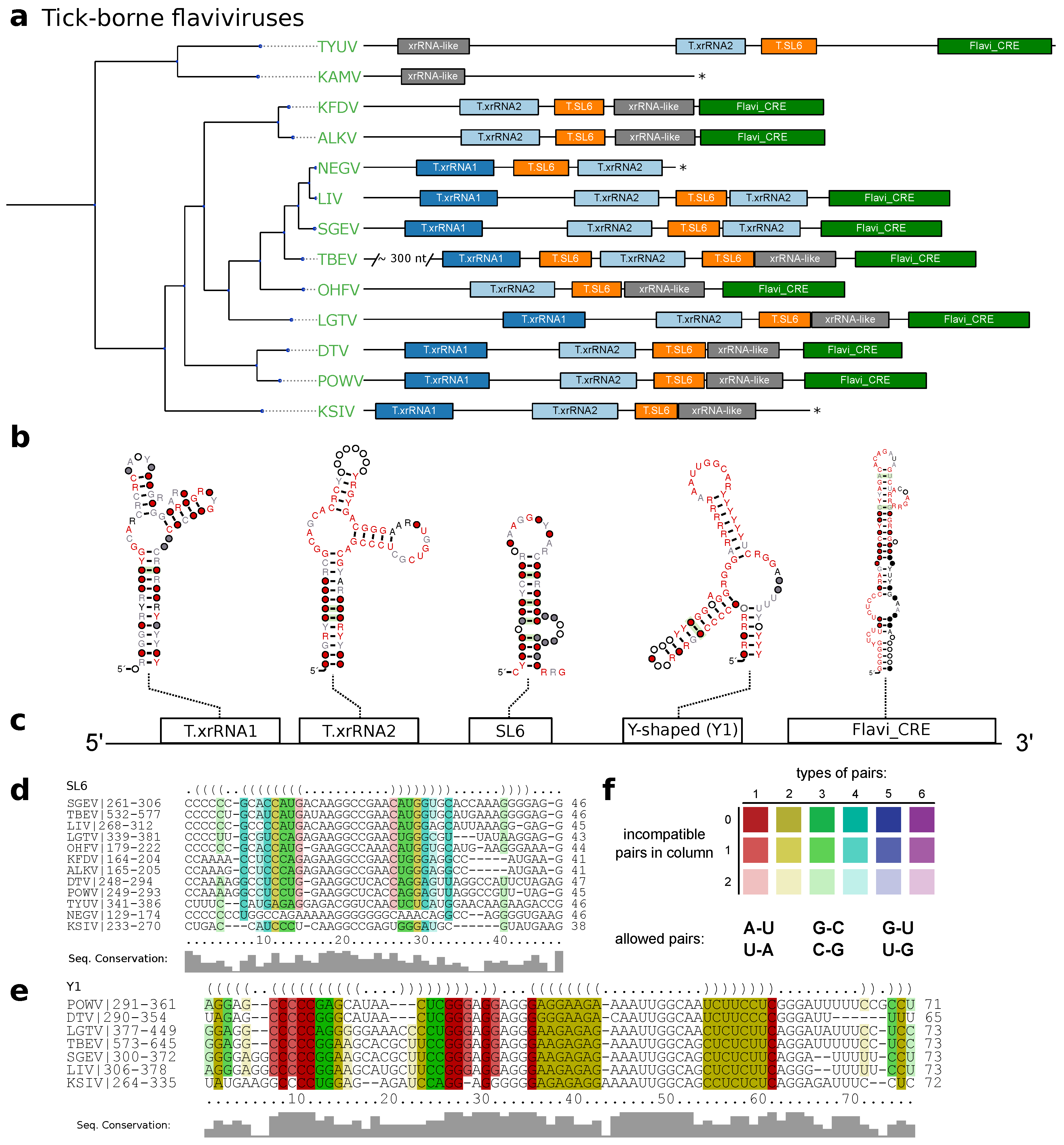
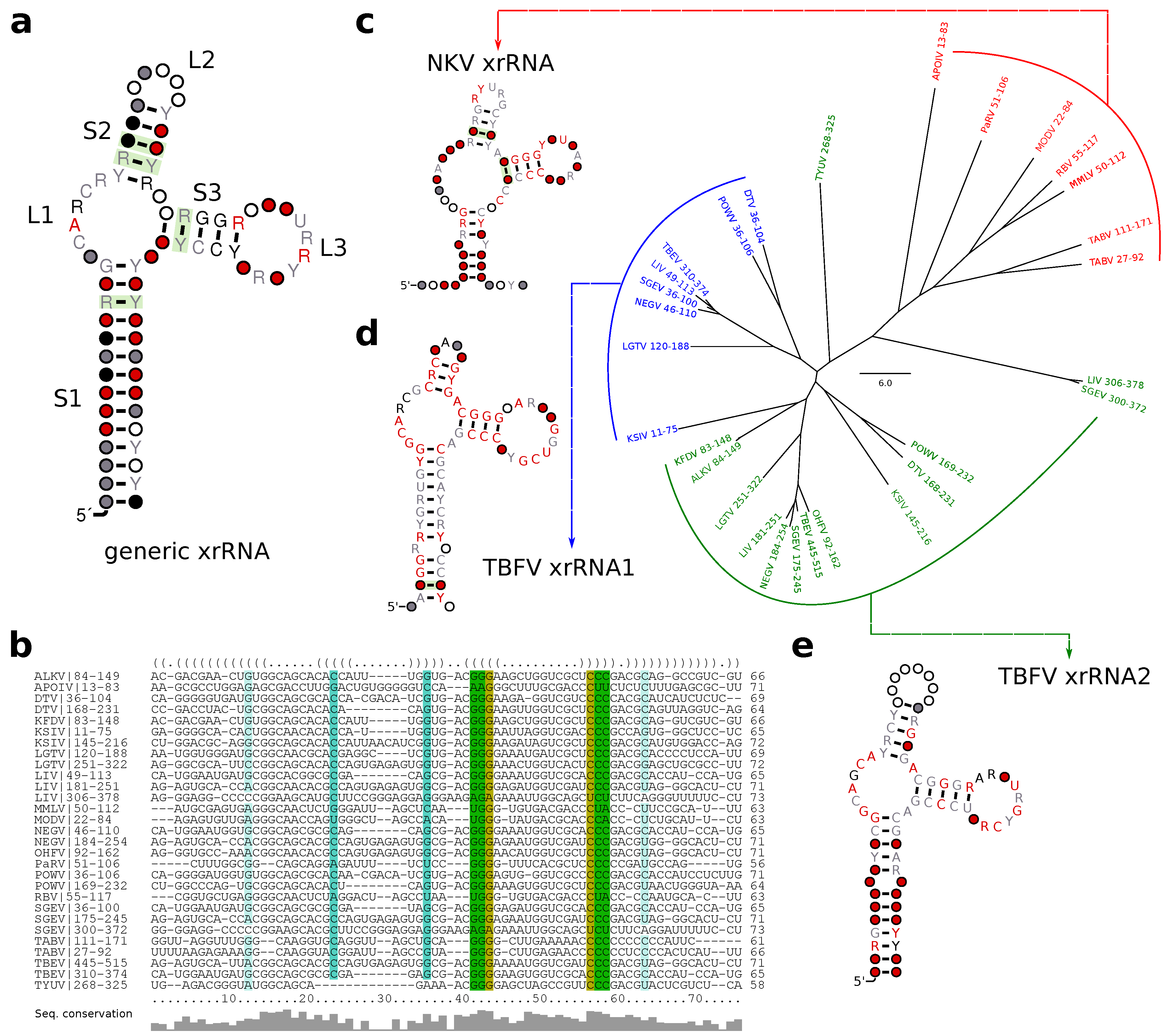
3.2. Tick-Borne Flaviviruses
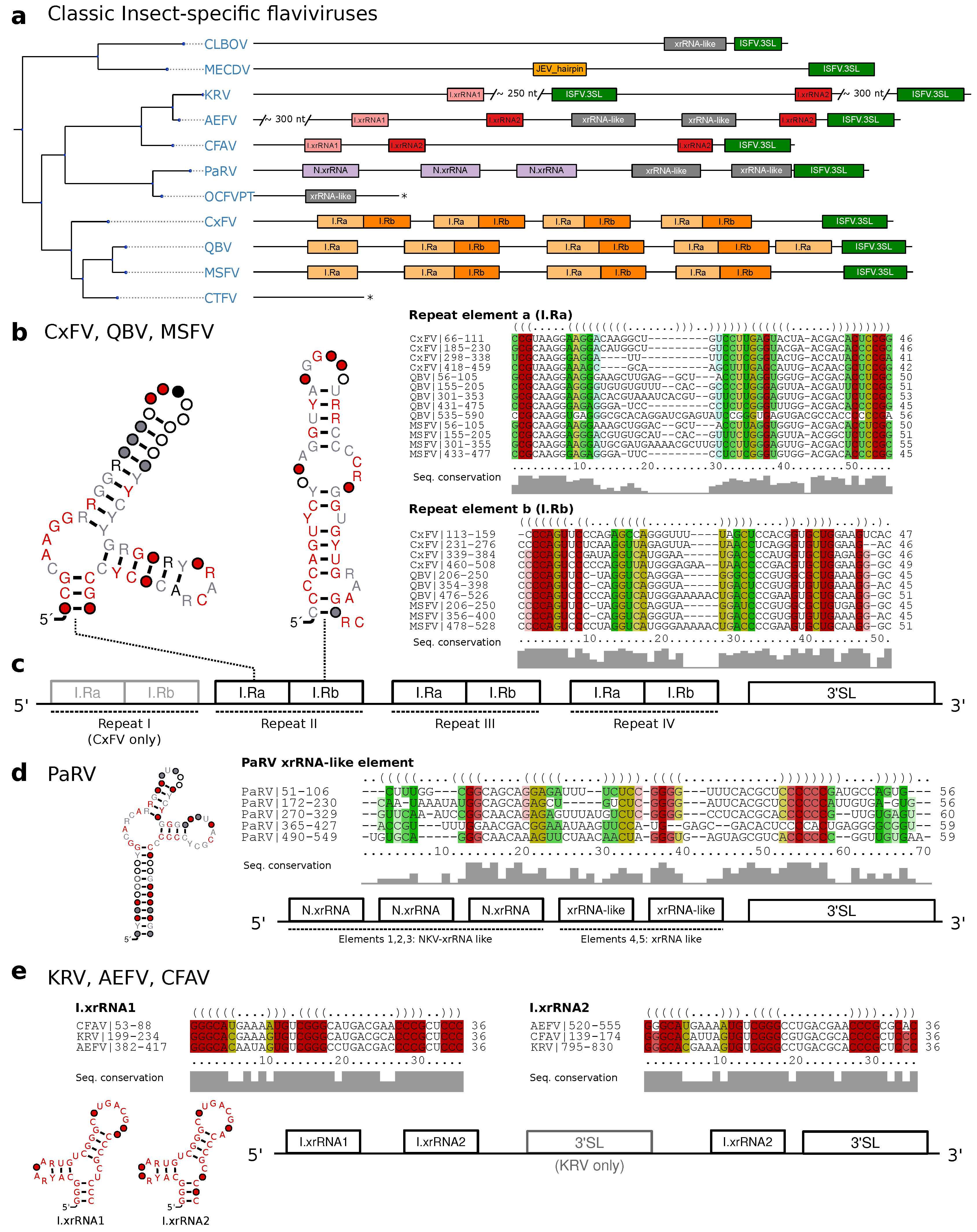
3.3. Classic Insect-Specific Flaviviruses
3.3.1. Exoribonuclease-Resistant RNAs in Aedes-Associated cISFVs
3.3.2. Conserved Structures in Culex-Associated cISFVs
3.3.3. Diverged 3UTR Architecture in Many cISFVs
3.4. Dual-Host Affiliated Insect-Specific Flaviviruses
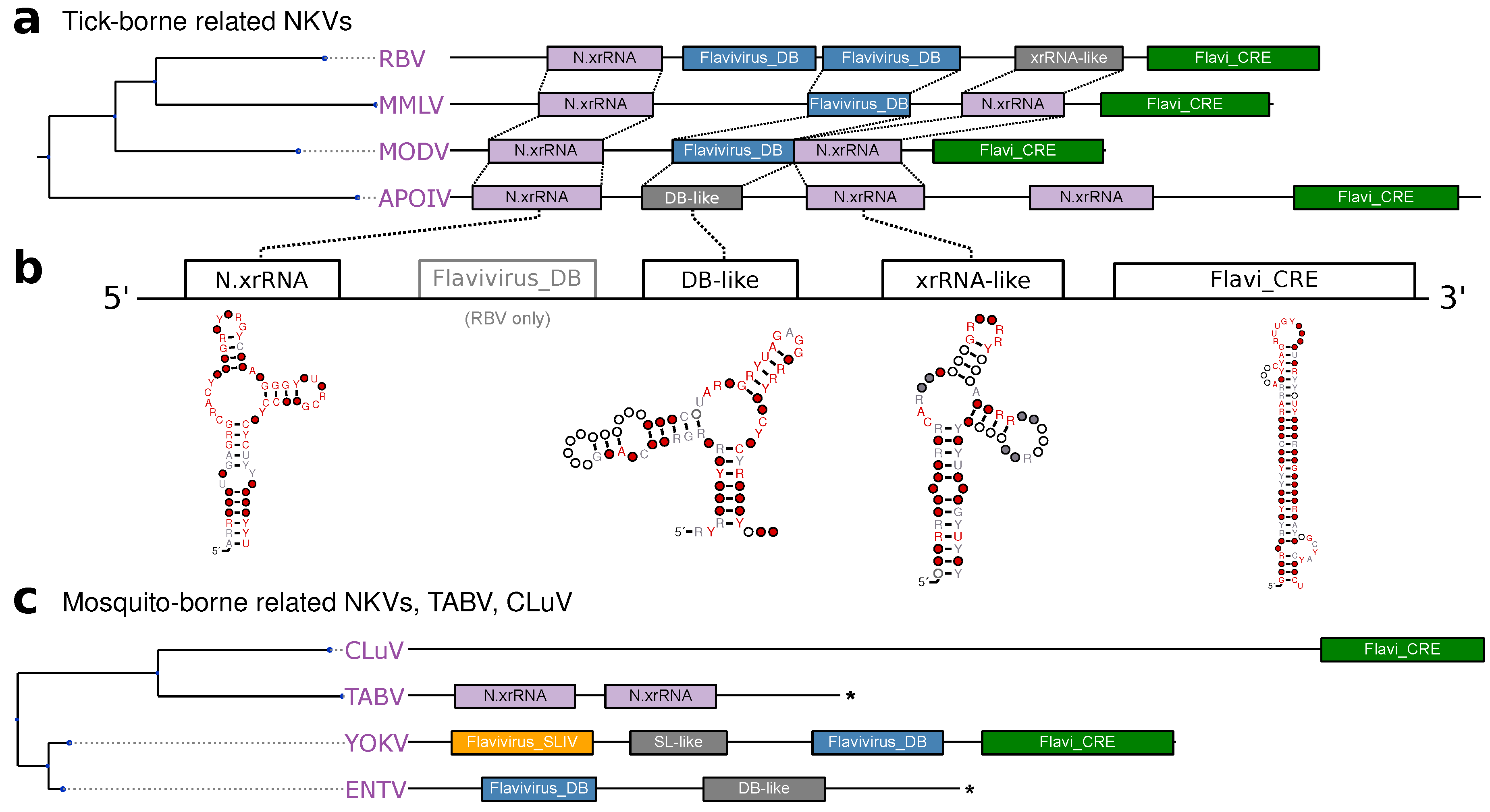
3.5. No-Known-Vector Flaviviruses
3.6. A Generalized xrRNA Structure
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Appendix A. Data Set Analyzed in This Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Accession Number | Acronym | Scientific Name | 3UTR Length (nt) | Isolates |
|---|---|---|---|---|---|
| ISFV | NC_012932.1 | AEFV | Aedes flavivirus | 942 | 3 |
| ISFV | NC_001564.2 | CFAV | Cell fusing agent virus | 553 | 3 |
| ISFV | KX669689.1 | CLBOV | Calbertado virus | 546 | 8 |
| ISFV | MF153378.1 | CTFV | Culex theileri flavivirus | 112 | 1 |
| ISFV | NC_008604.2 | CxFV | Culex flavivirus | 654 | 13 |
| ISFV | NC_030401.1 | HANV | Hanko virus | ||
| ISFV | NC_005064.1 | KRV | Kamiti River virus | 1208 | 3 |
| ISFV | NC_027819.1 | MECDV | Mercadeo virus | 638 | 3 |
| ISFV | NC_021069.1 | MSFV | Mosquito flavivirus | 674 | 6 |
| ISFV | NC_034242.1 | OCFVPT | Ochlerotatus caspius flavivirus | 148 | 2 |
| ISFV | NC_027817.1 | PaRV | Parramatta River virus | 629 | 2 |
| ISFV | NC_033694.1 | PCV | Palm Creek virus | ||
| ISFV | NC_012671.1 | QBV | Quang Binh virus | 673 | 2 |
| ISFV | MG214905.1 | BJV | Barkedji virus | 335 | 1 |
| ISFV | NC_017086.1 | CHAOV | Chaoyang virus | 326 | 2 |
| ISFV | NC_016997.1 | DONV | Donggang virus | 343 | 2 |
| ISFV | NC_027999.1 | EPEV | Paraiso Escondido virus | 316 | 2 |
| ISFV | NC_024805.1 | ILV | Ilomantsi virus | ||
| ISFV | KY320648.1 | KPKV | Kampung Karu virus | ||
| ISFV | FJ606789.2 | LAMV | Lammi virus | 326 | 1 |
| ISFV | KY290249.1 | LPKV | Long Pine Key virus | ||
| ISFV | KY320649.1 | LTNV | La Tina virus | ||
| ISFV | MF139576.1 | MMV | Marisma mosquito virus | 376 | 1 |
| ISFV | MF139575.1 | NANV | Nanay virus | 399 | 1 |
| ISFV | NC_024017.1 | NHUV | Nhumirim virus | 451 | 1 |
| ISFV | NC_033715.1 | NOUV | Nounane virus | 347 | 3 |
| NKV e | NC_003676.1 | APOIV | Apoi virus | 576 | 1 |
| NKV | KJ469370.1 | BCV | Batu Cave virus | ||
| NKV | MF776369.1 | CLuV | Cyclopterus lumpus virus | 601 | 1 |
| NKV | NC_008718.1 | ENTV | Entebbe bat virus | 308 | 3 |
| NKV e | NC_026620.1 | JUTV | Jutiapa virus | ||
| NKV | NC_004119.1 | MMLV | Montana myotis leukoencephalitis virus | 460 | 1 |
| NKV e | NC_003635.1 | MODV | Modoc virus | 366 | 1 |
| NKV | NC_034007.1 | PPBV | Phnom Penh bat virus | ||
| NKV | NC_003675.1 | RBV | Rio Bravo virus | 486 | 2 |
| NKV | NC_026624.1 | SOKV | Sokoluk virus | ||
| NKV | NC_003996.1 | TABV | Tamana bat virus | 241 | 1 |
| NKV | NC_005039.1 | YOKV | Yokose virus | 429 | 1 |
| TBFV | NC_004355.1 | ALKV | Alkhumra hemorrhagic fever virus | 393 | 21 |
| TBFV | AF311056.1 | DTV | Deer tick virus | 459 | 1 |
| TBFV | NC_033723.1 | GGV | Gadgets Gully virus | ||
| TBFV | NC_033724.1 | KADV | Kadam virus | ||
| TBFV | NC_023439.1 | KAMV | Kama virus | 282 | 2 |
| TBFV | HM055369.1 | KFDV | Kyasanur forest disease virus | 392 | 6 |
| TBFV | NC_006947.1 | KSIV | Karshi virus | 381 | 3 |
| TBFV | NC_003690.1 | LGTV | Langat virus | 568 | 5 |
| TBFV | NC_001809.1 | LIV | Louping ill virus | 500 | 5 |
| TBFV | NC_033721.1 | MEAV | Meaban virus | ||
| TBFV | KT224355.1 | NEGV | Negishi virus | 266 | 1 |
| TBFV | NC_005062.1 | OHFV | Omsk hemorrhagic fever virus | 410 | 4 |
| TBFV | NC_003687.1 | POWV | Powassan virus | 480 | 23 |
| TBFV | DQ235149.1 | RFV | Royal Farm virus | ||
| TBFV | NC_027709.1 | SGEV | Spanish goat encephalitis virus | 493 | 2 |
| TBFV | NC_033726.1 | SREV | Saumarez Reef virus | ||
| TBFV | NC_001672.1 | TBEV | Tick-borne encephalitis virus | 764 | 167 |
| TBFV | NC_023424.1 | TYUV | Tyuleniy virus | 591 | 3 |
References
- Kuno, G.; Chang, G.J.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the Genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar]
- Gaunt, M.W.; Sall, A.A.; de Lamballerie, X.; Falconar, A.K.; Dzhivanian, T.I.; Gould, E.A. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J. Gen. Virol. 2001, 82, 1867–1876. [Google Scholar] [CrossRef]
- Vasilakis, N.; Weaver, S.C. Flavivirus transmission focusing on Zika. Curr. Opin. Virol. 2017, 22, 30–35. [Google Scholar] [CrossRef]
- Barrows, N.J.; Campos, R.K.; Liao, K.C.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.C.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef]
- Kellman, E.; Offerdahl, D.; Melik, W.; Bloom, M. Viral Determinants of Virulence in Tick-Borne Flaviviruses. Viruses 2018, 10, 329. [Google Scholar] [CrossRef] [PubMed]
- Braack, L.; de Almeida, A.P.G.; Cornel, A.J.; Swanepoel, R.; De Jager, C. Mosquito-borne arboviruses of African origin: Review of key viruses and vectors. Parasite Vector 2018, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.; Lashkevich, V.; Gould, E. Tick-borne encephalitis. Antivir. Res. 2003, 57, 129–146. [Google Scholar] [CrossRef]
- Guzman, H.; Contreras-Gutierrez, M.A.; da Rosa, A.P.T.; Nunes, M.R.; Cardoso, J.F.; Popov, V.L.; Young, K.I.; Savit, C.; Wood, T.G.; Widen, S.G.; et al. Characterization of three new insect-specific flaviviruses: Their relationship to the mosquito-borne flavivirus pathogens. Am. J. Trop. Med. Hyg. 2018, 98, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Blitvich, B.; Firth, A. Insect-Specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Zapata, S.; Bichaud, L.; Moureau, G.; Lemey, P.; Firth, A.E.; Gritsun, T.S.; Gould, E.A.; de Lamballerie, X.; Depaquit, J.; et al. Ecuador Paraiso Escondido virus, a new flavivirus isolated from New World sandflies in Ecuador is the first representative of a novel clade in the genus Flavivirus. J. Virol. 2015, 89, 11773–11785. [Google Scholar] [CrossRef] [PubMed]
- Halbach, R.; Junglen, S.; van Rij, R.P. Mosquito-specific and mosquito-borne viruses: Evolution, infection, and host defense. Curr. Opin. Insect. Sci. 2017, 22, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.; Weaver, S.; Tesh, R.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef]
- Blitvich, B.; Firth, A. A Review of Flaviviruses that Have No Known Arthropod Vector. Viruses 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A.V. A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. Gene Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.E.; Ezcurra, A.L.D.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′ UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Reichert, E.D.; Polo, S.; Falgout, B.; Kasprzak, W.; Shapiro, B.A.; Padmanabhan, R. Identification of Cis-Acting Elements in the 3′-Untranslated Region of the Dengue Virus Type 2 RNA That Modulate Translation and Replication. J Biol. Chem. 2011, 286, 22521–22534. [Google Scholar] [CrossRef]
- Brinton, M.A.; Basu, M. Functions of the 3′ and 5′ genome RNA regions of members of the genus Flavivirus. Virus Res. 2015, 206, 108–119. [Google Scholar] [CrossRef]
- Ng, W.; Soto-Acosta, R.; Bradrick, S.; Garcia-Blanco, M.; Ooi, E. The 5′ and 3′ untranslated regions of the flaviviral genome. Viruses 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef] [PubMed]
- de Borba, L.; Villordo, S.M.; Marsico, F.L.; Carballeda, J.M.; Filomatori, C.V.; Gebhard, L.G.; Pallarés, H.M.; Lequime, S.; Lambrechts, L.; Vargas, I.S.; et al. RNA Structure Duplication in the Dengue Virus 3′ UTR: Redundancy or Host Specificity? mBio 2019, 10, e02506–18. [Google Scholar] [CrossRef]
- de Bernardi Schneider, A.; Wolfinger, M.T. Musashi binding elements in Zika and related Flavivirus 3′UTRs: A comparative study in silico. bioRxiv 2018. [Google Scholar] [CrossRef]
- Rice, C.M.; Lenches, E.M.; Eddy, S.R.; Shin, S.; Sheets, R.; Strauss, J. Nucleotide sequence of yellow fever virus: Implications for flavivirus gene expression and evolution. Science 1985, 229, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.S.; Hahn, Y.S.; Rice, C.M.; Lee, E.; Dalgarno, L.; Strauss, E.G.; Strauss, J.H. Conserved elements in the 3′ untranslated region of flavivirus RNAs and potential cyclization sequences. J. Mol. Biol. 1987, 198, 33–41. [Google Scholar] [CrossRef]
- Villordo, S.M.; Alvarez, D.E.; Gamarnik, A.V. A balance between circular and linear forms of the dengue virus genome is crucial for viral replication. RNA 2010, 16, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- de Borba, L.; Villordo, S.M.; Iglesias, N.G.; Filomatori, C.V.; Gebhard, L.G.; Gamarnik, A.V. Overlapping local and long-range RNA-RNA interactions modulate dengue virus genome cyclization and replication. J. Virol. 2015, 89, 3430–3437. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Funk, A.; Kondratieva, N.; Leung, J.; Torres, S.; Van der Aa, L.; Liu, W.J.; Palmenberg, A.C.; Shi, P.Y.; Hall, R.A.; et al. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 2008, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, B.M.; Eiler, D.; Kieft, J.S. Structured RNAs that evade or confound exonucleases: Function follows form. Curr. Opin. Struct. Biol. 2016, 36, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.L.; Anderson, J.R.; Kumagai, Y.; Wilusz, C.J.; Akira, S.; Khromykh, A.A.; Wilusz, J. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 2012, 18, 2029–2040. [Google Scholar] [CrossRef]
- Roby, J.A.; Pijlman, G.P.; Wilusz, J.; Khromykh, A.A. Noncoding subgenomic flavivirus RNA: Multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 2014, 6, 404–427. [Google Scholar] [CrossRef]
- Jones, C.I.; Zabolotskaya, M.V.; Newbury, S.F. The 5′-> 3′ exoribonuclease Xrn1/Pacman and its functions in cellular processes and development. Wiley Interdiscip. Rev. RNA 2012, 3, 455–468. [Google Scholar] [CrossRef]
- Antic, S.; Wolfinger, M.T.; Skucha, A.; Hosiner, S.; Dorner, S. General and miRNA-mediated mRNA degradation occurs on ribosome complexes in Drosophila cells. Mol. Cell Biol. 2015. [Google Scholar] [CrossRef]
- Kieft, J.S.; Rabe, J.L.; Chapman, E.G. New hypotheses derived from the structure of a flaviviral Xrn1-resistant RNA: Conservation, folding, and host adaptation. RNA Biol. 2015, 12, 1169–1177. [Google Scholar] [CrossRef]
- MacFadden, A.; O’Donoghue, Z.; Silva, P.A.; Chapman, E.G.; Olsthoorn, R.C.; Sterken, M.G.; Pijlman, G.P.; Bredenbeek, P.J.; Kieft, J.S. Mechanism and structural diversity of exoribonuclease-resistant RNA structures in flaviviral RNAs. Nat. Commun. 2018, 9, 119. [Google Scholar] [CrossRef]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; Khromykh, A.A.; Asgari, S. West Nile virus encodes a microRNA-like small RNA in the 3′ untranslated region which up-regulates GATA4 mRNA and facilitates virus replication in mosquito cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, A.; Funk, A.; Lazear, H.M.; Cooper, D.A.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef] [PubMed]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog. 2014, 10, e1004242. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Wolfinger, M.T.; Tanzer, A.; Hofacker, I.L. Predicting RNA Structures from Sequence and Probing Data. Methods 2016, 103, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.; Truong, K.; Nagasaki, T.; Torres, S.; Floden, N.; Melian, E.B.; Edmonds, J.; Dong, H.; Shi, P.Y.; Khromykh, A.A. RNA structures required for production of subgenomic flavivirus RNA. J. Virol. 2010, 84, 11407–11417. [Google Scholar] [CrossRef]
- Silva, P.A.; Pereira, C.F.; Dalebout, T.J.; Spaan, W.J.; Bredenbeek, P.J. An RNA pseudoknot is required for production of yellow fever virus subgenomic RNA by the host nuclease XRN1. J. Virol. 2010, 84, 11395–11406. [Google Scholar] [CrossRef] [PubMed]
- Sztuba-Solinska, J.; Teramoto, T.; Rausch, J.W.; Shapiro, B.A.; Padmanabhan, R.; Le Grice, S.F. Structural complexity of Dengue virus untranslated regions: cis-acting RNA motifs and pseudoknot interactions modulating functionality of the viral genome. Nucleic Acids Res. 2013, 41, 5075–5089. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.G.; Costantino, D.A.; Rabe, J.L.; Moon, S.L.; Wilusz, J.; Nix, J.C.; Kieft, J.S. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 2014, 344, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, B.M.; Laurence, H.M.; Massey, A.R.; Costantino, D.A.; Xie, X.; Yang, Y.; Shi, P.Y.; Nix, J.C.; Beckham, J.D.; Kieft, J.S. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 2016, 354, 1148–1152. [Google Scholar] [CrossRef]
- Rauscher, S.; Flamm, C.; Mandl, C.W.; Heinz, F.X.; Stadler, P.F. Secondary structure of the 3′-noncoding region of flavivirus genomes: comparative analysis of base pairing probabilities. RNA 1997, 3, 779–791. [Google Scholar] [PubMed]
- Hofacker, I.L.; Fekete, M.; Flamm, C.; Huynen, M.A.; Rauscher, S.; Stolorz, P.E.; Stadler, P.F. Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acid Res. 1998, 26, 3825–3836. [Google Scholar] [CrossRef] [PubMed]
- Witwer, C.; Rauscher, S.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Picornaviridae genomes. Nucleic Acid Res. 2001, 29, 5079–5089. [Google Scholar] [CrossRef]
- Hofacker, I.L.; Stadler, P.F.; Stocsits, R.R. Conserved RNA secondary structures in viral genomes: A survey. Bioinformatics 2004, 20, 1495–1499. [Google Scholar] [CrossRef]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold Faster RNA Homology Searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Waldl, M.; Thiel, B.C.; Ochsenreiter, R.; Holzenleiter, A.; de Araujo Oliveira, J.V.; Walter, M.E.M.; Wolfinger, M.T.; Stadler, P.F. TERribly Difficult: Searching for Telomerase RNAs in Saccharomycetes. Genes 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Will, S.; Reiche, K.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comp. Biol. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Bernhart, S.H.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithm Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Wolfinger, M.T.; Fallmann, J.; Eggenhofer, F.; Amman, F. ViennaNGS: A toolbox for building efficient next-generation sequencing analysis pipelines. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Wolfinger, M.T. Bio::RNA::RNAaliSplit 0.09. 2019. Available online: https://github.com/mtw/Bio-RNA-RNAaliSplit (accessed on 23 March 2019).
- Weinberg, Z.; Breaker, R.R. R2R-software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinform. 2011, 12, 3. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Gritsun, T.; Gould, E. Origin and Evolution of 3′UTR of Flaviviruses: Long Direct Repeats as A Basis for the Formation of Secondary Structures and Their Significance for Virus Transmission. Adv. Virus Res. 2006, 69, 203–248. [Google Scholar]
- Gritsun, D.J.; Jones, I.M.; Gould, E.A.; Gritsun, T.S. Molecular Archaeology of Flaviviridae Untranslated Regions: Duplicated RNA Structures in the Replication Enhancer of Flaviviruses and Pestiviruses Emerged via Convergent Evolution. PLoS ONE 2014, 9, e92056. [Google Scholar] [CrossRef]
- Gritsun, T.; Gould, E. The 3′ untranslated regions of Kamiti River virus and Cell fusing agent virus originated by self-duplication. J. Gen. Virol. 2006, 87, 2615–2619. [Google Scholar] [CrossRef]
- Hoshino, K.; Isawa, H.; Tsuda, Y.; Yano, K.; Sasaki, T.; Yuda, M.; Takasaki, T.; Kobayashi, M.; Sawabe, K. Genetic characterization of a new insect flavivirus isolated from Culex pipiens mosquito in Japan. Virology 2007, 359, 405–414. [Google Scholar] [CrossRef]
- Clarke, B.; Roby, J.; Slonchak, A.; Khromykh, A. Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res. 2015, 206, 53–61. [Google Scholar] [CrossRef]
- Moon, S.L.; Blackinton, J.G.; Anderson, J.R.; Dozier, M.K.; Dodd, B.J.; Keene, J.D.; Wilusz, C.J.; Bradrick, S.S.; Wilusz, J. X1 Stalling in the 5’UTR of Hepatitis C Virus and Bovine Viral Diarrhea Virus Is Associated with Dysregulated Host mRNA Stability. PLoS Pathog. 2015, 11, e1004708. [Google Scholar] [CrossRef]
- Charley, P.A.; Wilusz, C.J.; Wilusz, J. Identification of Phlebovirus and Arenavirus RNA sequences that stall and repress the exoribonuclease XRN1. J. Biol. Chem. 2018, 293, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Steckelberg, A.L.; Akiyama, B.M.; Costantino, D.A.; Sit, T.L.; Nix, J.C.; Kieft, J.S. A folded viral noncoding RNA blocks host cell exoribonucleases through a conformationally dynamic RNA structure. Proc. Natl. Acad. Sci. USA 2018, 115, 6404–6409. [Google Scholar] [CrossRef] [PubMed]
- Steckelberg, A.L.; Vicens, Q.; Kieft, J.S. Exoribonuclease-Resistant RNAs Exist within both Coding and Noncoding Subgenomic RNAs. mBio 2018, 9, e02461-18. [Google Scholar] [CrossRef] [PubMed]
- Iwakawa, H.o.; Mizumoto, H.; Nagano, H.; Imoto, Y.; Takigawa, K.; Sarawaneeyaruk, S.; Kaido, M.; Mise, K.; Okuno, T. A viral noncoding RNA generated by cis-element-mediated protection against 5′- 3′ RNA decay represses both cap-independent and cap-dependent translation. J. Virol. 2008, 82, 10162–10174. [Google Scholar] [CrossRef] [PubMed]
- Flobinus, A.; Hleibieh, K.; Klein, E.; Ratti, C.; Bouzoubaa, S.; Gilmer, D. A viral noncoding RNA complements a weakened viral RNA silencing suppressor and promotes efficient systemic host infection. Viruses 2016, 8, 272. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochsenreiter, R.; Hofacker, I.L.; Wolfinger, M.T. Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses 2019, 11, 298. https://doi.org/10.3390/v11030298
Ochsenreiter R, Hofacker IL, Wolfinger MT. Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses. 2019; 11(3):298. https://doi.org/10.3390/v11030298
Chicago/Turabian StyleOchsenreiter, Roman, Ivo L. Hofacker, and Michael T. Wolfinger. 2019. "Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses" Viruses 11, no. 3: 298. https://doi.org/10.3390/v11030298
APA StyleOchsenreiter, R., Hofacker, I. L., & Wolfinger, M. T. (2019). Functional RNA Structures in the 3′UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses. Viruses, 11(3), 298. https://doi.org/10.3390/v11030298