Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus


Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
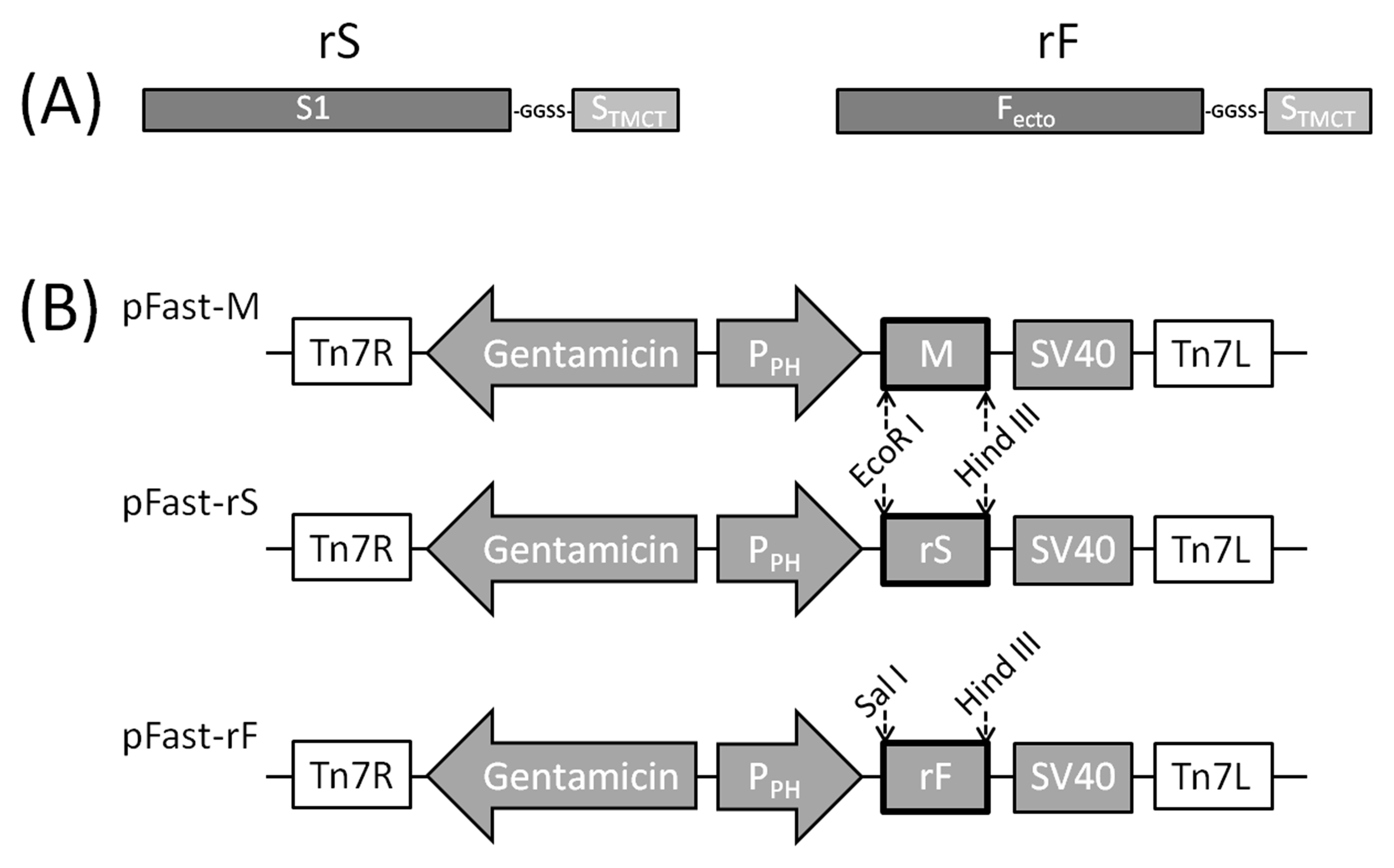
2.2. Construction of rS and rF Genes
2.3. Construction of Recombinant Baculoviruses Expressing rS, rF and M Genes
2.4. Western Blot Analysis of Protein Expression
2.5. Production and Purification of VLPs
2.6. Immunization and Challenge
2.7. Sample Collection
2.8. Evaluation of Vaccine Efficacy
2.9. Statistical Analysis
3. Results
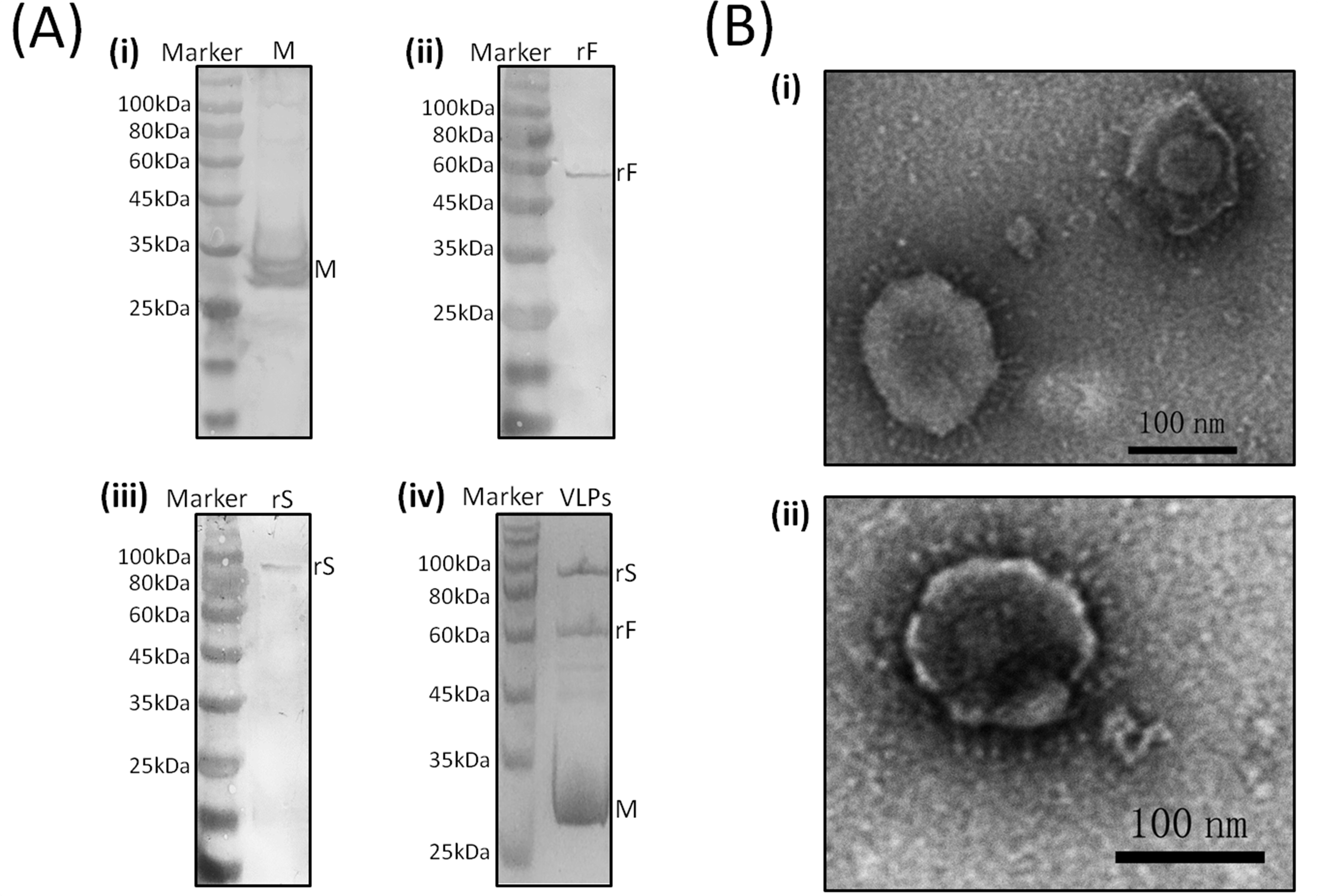
3.1. Western Blot Analyses of rS, rF, and M Protein Expression
3.2. Characterization of Chimeric IB-ND VLPs
3.3. Evaluation of IBV- and NDV-Specific Antibodies
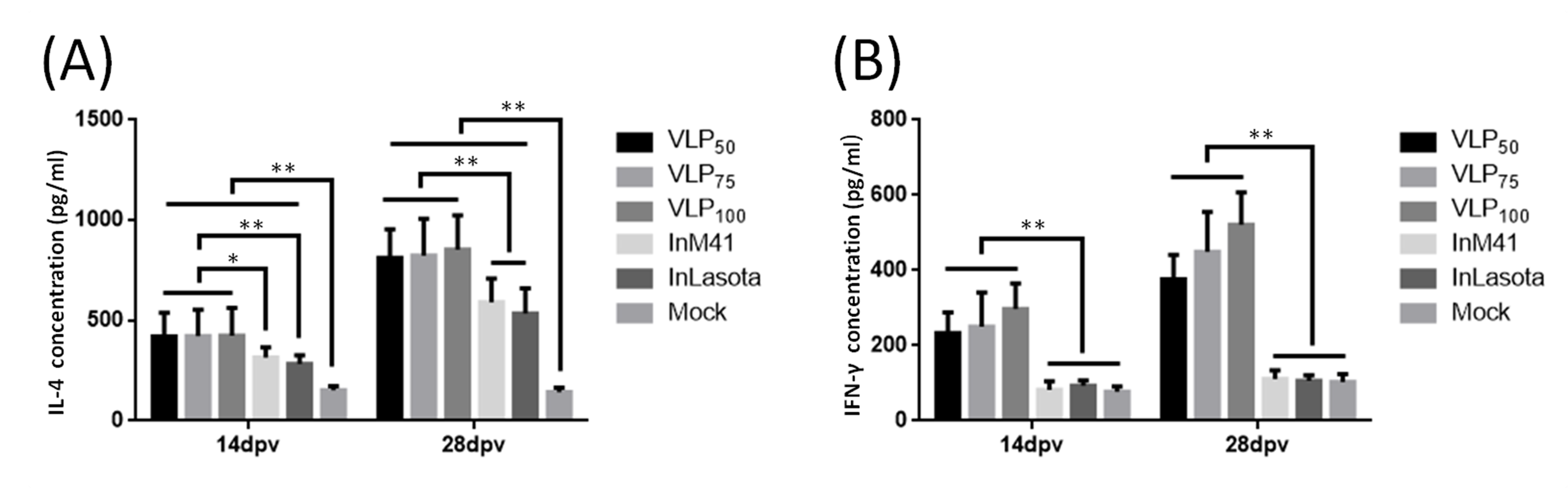
3.4. Cytokines Induction in Serum of Immunized Chickens
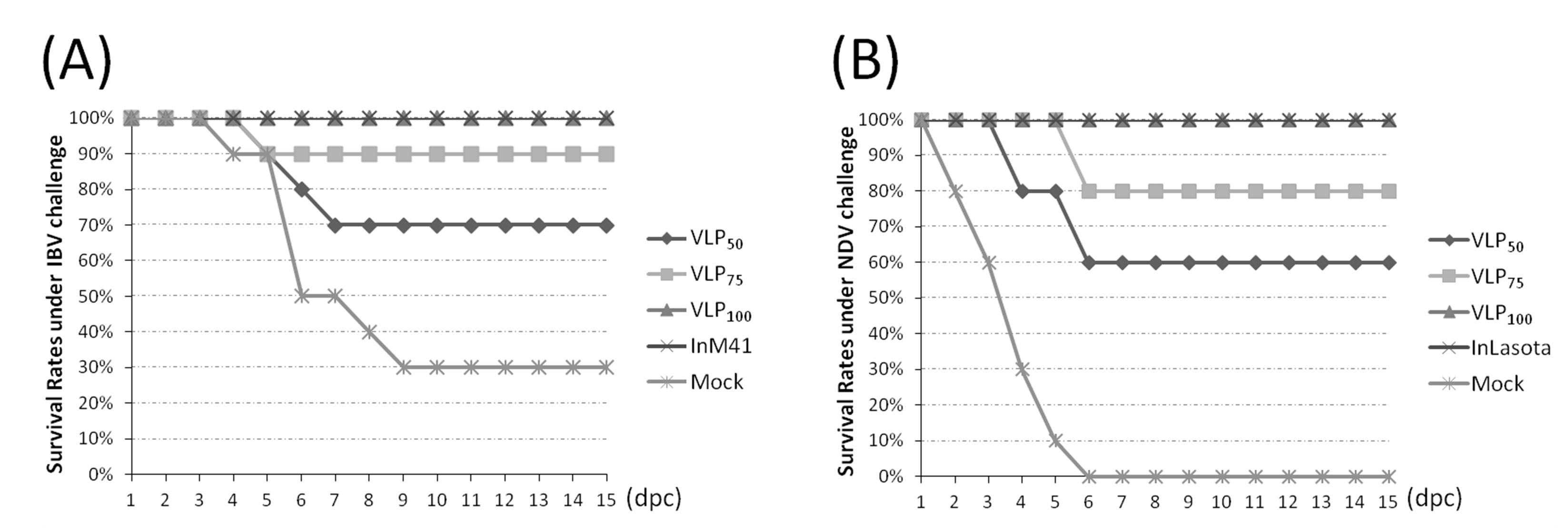
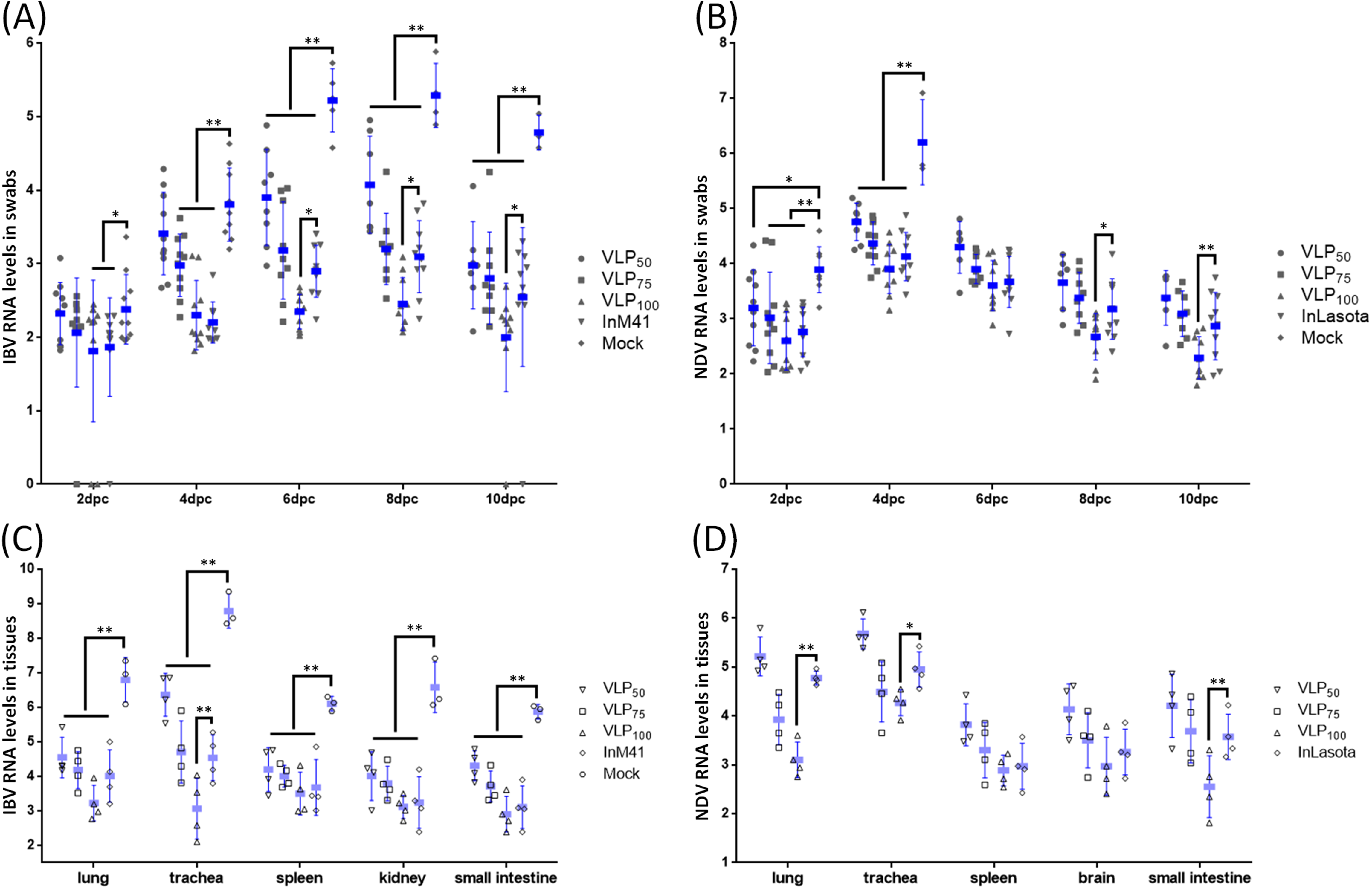
3.5. Evaluation of Protection Against IBV or NDV Challenge
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Ganar, K.; Das, M.; Sinha, S.; Kumar, S. Newcastle disease virus: Current status and our understanding. Virus Res. 2014, 184, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Godeke, G.J.; de Haan, C.A.M.; Rossen, J.W.A.; Vennema, H.; Rottier, P.J.M. Assembly of spikes into coronavirus particles is mediated by the carboxy-terminal domain of the spike protein. J. Virol. 2000, 74, 1566–1571. [Google Scholar] [CrossRef]
- Masters, P.S.; Kuo, L.; Ye, R.; Hurst, K.R.; Koetzner, C.A.; Hsue, B. Genetic and molecular biological analysis of protein-protein interactions in coronavirus assembly. Adv. Exp. Med. Biol. 2006, 581, 163–173. [Google Scholar]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Schwegmann-Wessels, C.; Neumann, U.; Herrler, G. The spike protein of infectious bronchitis virus is retained intracellularly by a tyrosine motif. J. Virol. 2008, 82, 2765–2771. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Pappin, D.J.C.; Binns, M.M.; Boursnell, M.E.G.; Brown, T.D.K. Coronavirus IBV: Partial amino terminal sequencing of spike polypeptide S2 identifies the sequence Arg-Arg-Phe-Arg-Arg at the cleavage site of the spike precursor propolypeptide of IBV strains Beaudette and M41. Virus Res. 1986, 4, 133–143. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Darbyshire, J.H.; Peters, R.W. Coronavirus IBV: Virus retaining spike glycopolypeptide S2 but not S1 is unable to induce virus-neutralizing or haemagglutination-inhibiting antibody, or induce chicken tracheal protection. J. Gen. Virol. 1986, 67, 1435–1442. [Google Scholar] [CrossRef]
- de Leeuw, O.; Peeters, B. Complete nucleotide sequence of Newcastle disease virus: Evidence for the existence of a new genus within the subfamily Paramyxovirinae. J. Gen. Virol. 1999, 80, 131–136. [Google Scholar] [CrossRef]
- Panda, A.; Huang, Z.; Elankumaran, S.; Rockemann, D.D.; Samal, S.K. Role of fusion protein cleavage site in the virulence of Newcastle disease virus. Microb. Pathog. 2004, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- McGinnes, L.W.; Reitter, J.N.; Gravel, K.; Morrison, T.G. Evidence for Mixed Membrane Topology of the Newcastle Disease Virus Fusion Protein. J. Virol. 2003, 77, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Nakamura, H.; Sonoda, K.; Hamada, F.; Hirai, K. Protection of chickens from Newcastle disease by vaccination with a linear plasmid DNA expressing the F protein of Newcastle disease virus. Vaccine 1996, 14, 747–752. [Google Scholar] [CrossRef]
- Noh, J.Y.; Park, J.K.; Lee, D.H.; Yuk, S.S.; Kwon, J.H.; Lee, S.W.; Lee, J.B.; Park, S.Y.; Choi, I.S.; Song, C.S. Chimeric Bivalent Virus-Like Particle Vaccine for H5N1 HPAI and ND Confers Protection against a Lethal Challenge in Chickens and Allows a Strategy of Differentiating Infected from Vaccinated Animals (DIVA). PloS ONE 2016, 11, e0162946. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nayak, B.; Collins, P.L.; Samal, S.K. Evaluation of the Newcastle disease virus F and HN proteins in protective immunity by using a recombinant avian paramyxovirus type 3 vector in chickens. J. Virol. 2011, 85, 6521–6534. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Lee, D.H.; Yuk, S.S.; Tseren-Ochir, E.O.; Kwon, J.H.; Noh, J.Y.; Kim, B.Y.; Choi, S.W.; Kang, S.M.; Lee, J.B.; et al. Virus-like particle vaccine confers protection against a lethal newcastle disease virus challenge in chickens and allows a strategy of differentiating infected from vaccinated animals. Clin. Vaccine Immunol. CVI 2014, 21, 360–365. [Google Scholar] [CrossRef]
- Bande, F.; Arshad, S.S.; Bejo, M.H.; Moeini, H.; Omar, A.R. Progress and challenges toward the development of vaccines against avian infectious bronchitis. J. Immunol. Res. 2015, 2015, 424860. [Google Scholar] [CrossRef]
- Noad, R.; Roy, P. Virus-like particles as immunogens. Trends Microbiol. 2003, 11, 438–444. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Xu, P.; Zhou, L.; Zhang, Z.; Wang, H. Genome sequence and origin analyses of the recombinant novel IBV virulent isolate SAIBK2. Virus Genes 2016, 52, 509–520. [Google Scholar] [CrossRef]
- Zhang, T.; Han, Z.; Xu, Q.; Wang, Q.; Gao, M.; Wu, W.; Shao, Y.; Li, H.; Kong, X.; Liu, S. Serotype shift of a 793/B genotype infectious bronchitis coronavirus by natural recombination. Infect. Genet. Evol. 2015, 32, 377–387. [Google Scholar] [CrossRef]
- Chare, E.R.; Gould, E.A.; Holmes, E.C. Phylogenetic analysis reveals a low rate of homologous recombination in negative-sense RNA viruses. J. Gen. Virol. 2003, 84, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Sun, L.; Ma, B.; Cui, Z.; Zhu, Y.; Kitamura, Y.; Liu, W. F gene recombination between genotype II and VII Newcastle disease virus. Virus Res. 2008, 131, 299–303. [Google Scholar] [CrossRef]
- McGinnes, L.W.; Pantua, H.; Laliberte, J.P.; Gravel, K.A.; Jain, S.; Morrison, T.G. Assembly and biological and immunological properties of Newcastle disease virus-like particles. J. Virol. 2010, 84, 4513–4523. [Google Scholar] [CrossRef]
- Tan, M.; Jiang, X. Subviral particle as vaccine and vaccine platform. Curr. Opin. Virol. 2014, 6, 24–33. [Google Scholar] [CrossRef]
- Plummer, E.M.; Manchester, M. Viral nanoparticles and virus-like particles: Platforms for contemporary vaccine design. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2011, 3, 174–196. [Google Scholar] [CrossRef] [PubMed]
- Lua, L.H.L.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P.J. Bioengineering Virus-Like Particles as Vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef]
- Kushnir, N.; Streatfield, S.J.; Yusibov, V. Virus-like particles as a highly efficient vaccine platform: Diversity of targets and production systems and advances in clinical development. Vaccine 2012, 31, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.; Medina, A.; Rutter, W.J.; Ammerer, G.; Hall, B.D. Synthesis and assembly of hepatitis B virus surface antigen particles in yeast. Nature 1982, 298, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Lv, L.; Yin, L.; Li, X.; Luo, D.; Liu, K.; Xue, C.; Cao, Y. Assembly and immunogenicity of coronavirus-like particles carrying infectious bronchitis virus M and S proteins. Vaccine 2013, 31, 5524–5530. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.W.; Wu, X.; Wang, H.N.; Ma, B.C.; Ding, M.D.; Yang, X. Assembly and immunogenicity of baculovirus-derived infectious bronchitis virus-like particles carrying membrane, envelope and the recombinant spike proteins. Biotechnol. Lett. 2016, 38, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kong, X. A new genotype of nephropathogenic infectious bronchitis virus circulating in vaccinated and non-vaccinated flocks in China. Avian Pathol. 2004, 33, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Sun, J.; Qi, T.; Zhao, W.; Han, Z.; Yang, X.; Liu, S. Recombinant Newcastle disease virus expressing the infectious bronchitis virus S1 gene protects chickens against Newcastle disease virus and infectious bronchitis virus challenge. Vaccine 2017, 35, 2435–2442. [Google Scholar] [CrossRef]
- Toyoda, T.; Gotoh, B.; Sakaguchi, T.; Kida, H.; Nagai, Y. Identification of Amino Acids Relevant to Three Antigenic Determinants on the Fusion Protein of Newcastle Disease Virus That Are Involved in Fusion Inhibition and Neutralization. J. Virol. 1988, 62, 4427–4430. [Google Scholar] [PubMed]
- Hu, S.; Ma, H.; Wu, Y.; Liu, W.; Wang, X.; Liu, Y.; Liu, X. A vaccine candidate of attenuated genotype VII Newcastle disease virus generated by reverse genetics. Vaccine 2009, 27, 904–910. [Google Scholar] [CrossRef]
- Schijns, V.E. Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 2000, 12, 456–463. [Google Scholar] [CrossRef]
- Chen, L.; Yin, L.; Zhou, Q.; Li, Q.; Luo, Y.; Xu, Z.; Zhang, Y.; Xue, C.; Cao, Y. Immunogenicity and protective efficacy of recombinant fiber-2 protein in protecting SPF chickens against fowl adenovirus 4. Vaccine 2018, 36, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lai, H.; Sun, H.; Chen, Q. Virus-like particles that display Zika virus envelope protein domain III induce potent neutralizing immune responses in mice. Sci. Rep. 2017, 7, 7679. [Google Scholar] [CrossRef]
- Phoolcharoen, W.; Dye, J.M.; Kilbourne, J.; Piensook, K.; Pratt, W.D.; Arntzen, C.J.; Chen, Q.; Mason, H.S.; Herbst-Kralovetz, M.M. A nonreplicating subunit vaccine protects mice against lethal Ebola virus challenge. Proc. Natl. Acad. Sci. USA 2011, 108, 20695–20700. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.; Medzhitov, R. Toll-like receptors and their ligands. In Toll-Like Receptor Family Members and Their Ligands; Springer: Berlin, Germany, 2002; pp. 81–92. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′→3′) | Genomic Position of Primers (nt) | Length of Product (bp) |
|---|---|---|---|
| S1-F | GAATTCATGTTGGTAACACCTCTTTTACTAG | 20374—20398 | 1620 |
| S1-R | CGCGGATCCTCCACGACGTGTTCCATTA | 21975—21960 | |
| Fecto-F | GTCGACATGGGCTCCAGACCTTCTACC | 4544—4564 | 1515 |
| Fecto-R | CGCGGATCCTCCAGATGTGCTAGTCAG | 6040—6026 | |
| STMCT-F | CGCGGATCCTCATGGTATGTGTGG | 23653—23664 | 228 |
| STMCT-R | AAGCTTTTAAACAGACTTTTTAGGTCTG | 23862—23841 | |
| M-F | GAATTCATGTCCAACGAAACAAATTGTAC | 24511—24533 | 690 |
| M-R | AAGCTTTTATGTGTAAAGGCTACTTCCACCTG | 25188—25163 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Zhai, X.; Lai, Y.; Zuo, L.; Zhang, Y.; Mei, X.; Xiang, R.; Kang, Z.; Zhou, L.; Wang, H. Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus. Viruses 2019, 11, 254. https://doi.org/10.3390/v11030254
Wu X, Zhai X, Lai Y, Zuo L, Zhang Y, Mei X, Xiang R, Kang Z, Zhou L, Wang H. Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus. Viruses. 2019; 11(3):254. https://doi.org/10.3390/v11030254
Chicago/Turabian StyleWu, Xuan, Xiwen Zhai, Yan Lai, Lei Zuo, Yu Zhang, Xueran Mei, Rong Xiang, Zhuangzhuang Kang, Long Zhou, and Hongning Wang. 2019. "Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus" Viruses 11, no. 3: 254. https://doi.org/10.3390/v11030254
APA StyleWu, X., Zhai, X., Lai, Y., Zuo, L., Zhang, Y., Mei, X., Xiang, R., Kang, Z., Zhou, L., & Wang, H. (2019). Construction and Immunogenicity of Novel Chimeric Virus-Like Particles Bearing Antigens of Infectious Bronchitis Virus and Newcastle Disease Virus. Viruses, 11(3), 254. https://doi.org/10.3390/v11030254