Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes


Abstract
1. Introduction
2. Materials and Methods
2.1. Genomes Analyzed
2.2. Trna Gene Prediction, Identification, and Classification of tRNA Gene Clusters
2.3. Taxonomic Designation, Sequence Annotation, and Gene Content Analysis
2.4. Phylogenetic Analysis
2.5. Codon Bias and Comparative Analyzes of the tRNA Genes
2.6. Statistical Analysis
3. Results
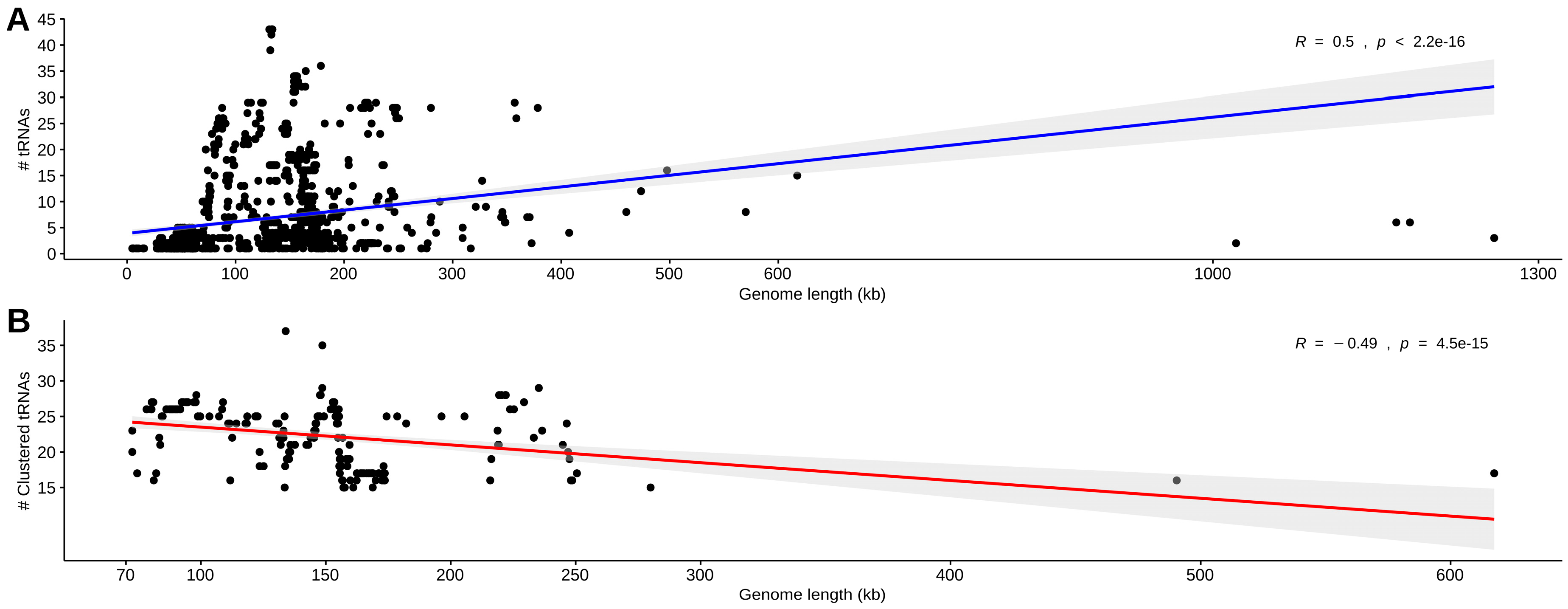
3.1. Data Set Classification and tRNA Gene Distribution
3.2. Identification, Characterization, and Organization of tRNA Gene Clusters in Phage and Virus Genomes
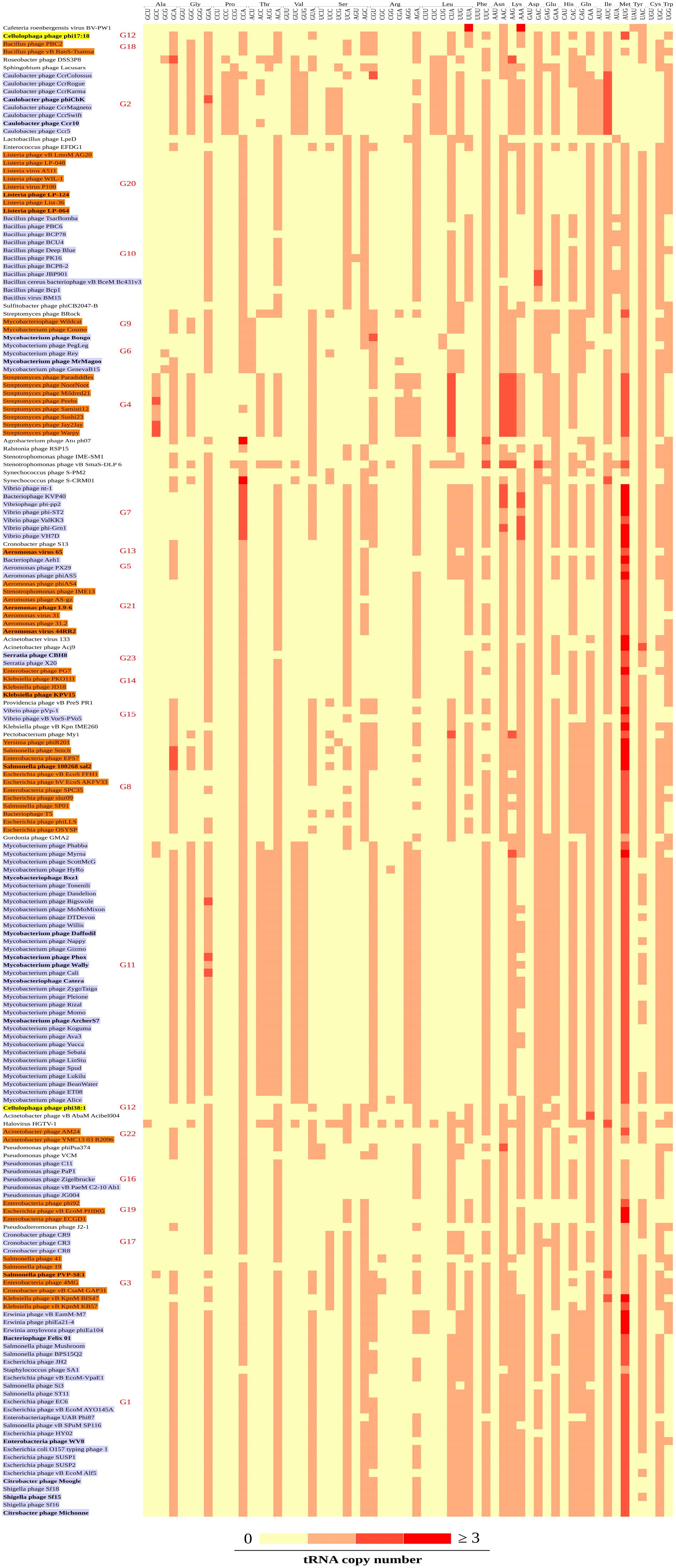
3.3. Codon Patterns in the tRNA Gene Clusters
3.4. CDS and tRNA Gene Cluster Groups
3.5. Source of the Phage tRNA Gene Clusters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef]
- Delesalle, V.A.; Tanke, N.T.; Vill, A.C.; Krukonis, G.P. Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 2016, 6, e1219441. [Google Scholar] [CrossRef]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef]
- Bowden, R.J.; Simas, J.P.; Davis, A.J.; Efstathiou, S. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol. 1997, 78 Pt 7, 1675–1687. [Google Scholar] [CrossRef]
- Amgarten, D.; Martins, L.F.; Lombardi, K.C.; Antunes, L.P.; de Souza, A.P.S.; Nicastro, G.G.; Kitajima, E.W.; Quaggio, R.B.; Upton, C.; Setubal, J.C.; et al. Three novel Pseudomonas phages isolated from composting provide insights into the evolution and diversity of tailed phages. BMC Genom. 2017, 18, 346. [Google Scholar] [CrossRef]
- Dreher, T.W. Viral tRNAs and tRNA-like structures. Wiley Interdiscip. Rev. RNA 2010, 1, 402–414. [Google Scholar] [CrossRef]
- Colson, P.; Fournous, G.; Diene, S.M.; Raoult, D. Codon usage, amino acid usage, transfer RNA and amino-acyl-tRNA synthetases in Mimiviruses. Intervirology 2013, 56, 364–375. [Google Scholar] [CrossRef]
- Albers, S.; Czech, A. Exploiting tRNAs to Boost Virulence. Life 2016, 6, 4. [Google Scholar] [CrossRef]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Jacobs-Sera, D.; Lawrence, J.G.; Pope, W.H.; Russell, D.A.; Ko, C.C.; Weber, R.J.; Patel, M.C.; Germane, K.L.; Edgar, R.H.; et al. Comparative genomic analysis of 60 Mycobacteriophage genomes: Genome clustering, gene acquisition, and gene size. J. Mol. Biol. 2010, 397, 119–143. [Google Scholar] [CrossRef]
- Senčilo, A.; Jacobs-Sera, D.; Russell, D.A.; Ko, C.C.; Bowman, C.A.; Atanasova, N.S.; Österlund, E.; Oksanen, H.M.; Bamford, D.H.; Hatfull, G.F.; et al. Snapshot of haloarchaeal tailed virus genomes. RNA Biol. 2013, 10, 803–816. [Google Scholar] [CrossRef]
- Pope, W.H.; Anders, K.R.; Baird, M.; Bowman, C.A.; Boyle, M.M.; Broussard, G.W.; Chow, T.; Clase, K.L.; Cooper, S.; Cornely, K.A.; et al. Cluster M mycobacteriophages Bongo, PegLeg, and Rey with unusually large repertoires of tRNA isotypes. J. Virol. 2014, 88, 2461–2480. [Google Scholar] [CrossRef]
- Morgado, S.M.; Vicente, A.C.P. Beyond the Limits: tRNA Array Units in Mycobacterium Genomes. Front. Microbiol. 2018, 9, 1042. [Google Scholar] [CrossRef]
- Jung, P.P.; Friedrich, A.; Souciet, J.L.; Louis, V.; Potier, S.; de Montigny, J.; Schacherer, J. Complete mitochondrial genome sequence of the yeast Pichia farinosa and comparative analysis of closely related species. Curr. Genet. 2010, 56, 507–515. [Google Scholar] [CrossRef]
- Friedrich, A.; Jung, P.P.; Hou, J.; Neuvéglise, C.; Schacherer, J. Comparative Mitochondrial Genomics within and among Yeast Species of the Lachancea Genus. PLoS ONE 2012, 7, e47834. [Google Scholar] [CrossRef]
- Li, E.; Li, X.; Wu, X.; Feng, G.; Zhang, M.; Shi, H.; Wang, L.; Jiang, J. Complete nucleotide sequence and gene rearrangement of the mitochondrial genome of Occidozyga martensii. J. Genet. 2014, 93, 631–641. [Google Scholar] [CrossRef]
- Morgado, S.M.; Vicente, A.C.P. Exploring tRNA gene cluster in archaea. Mem. Inst. Oswaldo Cruz 2019, 114, e180348. [Google Scholar] [CrossRef]
- Tawari, B.; Ali, I.K.; Scott, C.; Quail, M.A.; Berriman, M.; Hall, N.; Clark, C.G. Patterns of evolution in the unique tRNA gene arrays of the genus Entamoeba. Mol. Biol. Evol. 2008, 25, 187–198. [Google Scholar] [CrossRef]
- Bermudez-Santana, C.; Attolini, C.S.-O.; Kirsten, T.; Engelhardt, J.; Prohaska, S.J.; Steigele, S.; Stadler, P.F. Genomic organization of eukaryotic tRNAs. BMC Genom. 2010, 11, 270. [Google Scholar] [CrossRef]
- Puerto-Galan, L.; Vioque, A. Expression and processing of an unusual tRNA gene cluster in the cyanobacterium Anabaena sp. PCC 7120. FEMS Microbiol. Lett. 2012, 337, 10–17. [Google Scholar] [CrossRef]
- Tran, T.T.T.; Belahbib, H.; Bonnefoy, V.; Talla, E. A Comprehensive tRNA Genomic Survey Unravels the Evolutionary History of tRNA Arrays in Prokaryotes. Genome Biol. Evol. 2016, 8, 282–295. [Google Scholar] [CrossRef]
- Alamos, P.; Tello, M.; Bustamante, P.; Gutiérrez, F.; Shmaryahu, A.; Maldonado, J.; Levicán, G.; Orellana, O. Functionality of tRNAs encoded in a mobile genetic element from an acidophilic bacterium. RNA Biol. 2018, 15, 518–527. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Lanza, V.F.; Baquero, F.; de la Cruz, F.; Coque, T.M. AcCNET (Accessory Genome Constellation Network): Comparative genomics software for accessory genome analysis using bipartite networks. Bioinformatics 2017, 33, 283–285. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Abe, T.; Inokuchi, H.; Yamada, Y.; Muto, A.; Iwasaki, Y.; Ikemura, T. tRNADB-CE: tRNA gene database well-timed in the era of big sequence data. Front. Genet. 2014, 5, 114. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org (accessed on 31 December 2018).
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2015; Available online: http://www.rstudio.com (accessed on 31 December 2018).
- Hunnicutt, L.E.; Hunter, W.B.; Cave, R.D.; Powell, C.A.; Mozoruk, J.J. Genome sequence and molecular characterization of Homalodisca coagulata virus-1, a novel virus discovered in the glassy-winged sharpshooter (Hemiptera: Cicadellidae). Virology 2006, 350, 67–78. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Canuti, M.; Deijs, M.; de Vries, M.; Jebbink, M.F.; Rebers, S.; Molenkamp, R.; van Hemert, F.J.; Chung, K.; Cotten, M.; et al. Unexplained diarrhoea in HIV-1 infected individuals. BMC Infect. Dis. 2014, 14, 22. [Google Scholar] [CrossRef]
- Ibaba, J.D.; Laing, M.D.; Gubba, A. Pepo aphid-borne yellows virus: A new species inthe genus Polerovirus. Virus Genes. 2017, 53, 134–136. [Google Scholar] [CrossRef]
- Shirako, Y.; Wilson, T.M. Complete nucleotide sequence and organization of thebipartite RNA genome of soil-borne wheat mosaic virus. Virology 1993, 195, 16–32. [Google Scholar] [CrossRef]
- Chithambaram, S.; Prabhakaran, R.; Xia, X. Differential codon adaptation between dsDNA and ssDNA phages in Escherichia coli. Mol. Biol. Evol. 2014, 31, 1606–1617. [Google Scholar] [CrossRef]
- Rak, R.; Dahan, O.; Pilpel, Y. Repertoires of tRNAs: The Couplers of Genomics and Proteomics. Ann. Rev. Cell Dev. Biol. 2018, 34, 239–264. [Google Scholar] [CrossRef]
- Chopin, A.; Bolotin, A.; Sorokin, A.; Ehrlich, S.D.; Chopin, M.-C. Analysis of six prophages in Lactococcus lactis IL1403: Different genetic structure of temperate and virulent phage populations. Nucleic Acids Res. 2001, 29, 644–651. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017, 2, 17112. [Google Scholar] [CrossRef]
- Miller, E.S.; Heidelberg, J.F.; Eisen, J.A.; Nelson, W.C.; Durkin, A.S.; Ciecko, A.; Feldblyum, T.V.; White, O.; Paulsen, I.T.; Nierman, W.C.; et al. Complete genome sequence of the broad-host-range vibriophage KVP40: Comparative genomics of a T4-related bacteriophage. J. Bacteriol. 2003, 185, 5220–5233. [Google Scholar] [CrossRef]
- Lehman, S.M.; Kropinski, A.M.; Castle, A.J.; Svircev, A.M. Complete Genome of the Broad-Host-Range Erwinia amylovora Phage ΦEa21-4 and Its Relationship to Salmonella Phage Felix O1. Appl. Environ. Microbiol. 2009, 75, 2139–2147. [Google Scholar] [CrossRef]
- Santos, S.B.; Kropinski, A.M.; Ceyssens, P.J.; Ackermann, H.W.; Villegas, A.; Lavigne, R.; Krylov, V.N.; Carvalho, C.M.; Ferreira, E.C.; Azeredo, J. Genomic and proteomic characterization of the broad-host-range Salmonella phage PVP-SE1: Creation of a new phage genus. J. Virol. 2011, 85, 11265–11273. [Google Scholar] [CrossRef]
- Kim, J.H.; Son, J.S.; Choi, Y.J.; Choresca, C.H., Jr.; Shin, S.P.; Han, J.E.; Jun, J.W.; Park, S.C. Complete genome sequence and characterization of a broad-host range T4-like bacteriophage phiAS5 infecting Aeromonas salmonicida subsp. salmonicida. Vet. Microbiol. 2011, 157, 164–171. [Google Scholar] [CrossRef]
- Schwarzer, D.; Buettner, F.F.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A multivalent adsorption apparatus explains the broad host range of phage phi92: A comprehensive genomic and structural analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef]
- El-Arabi, T.F.; Griffiths, M.W.; She, Y.-M.; Villegas, A.; Lingohr, E.J.; Kropinski, A.M. Genome sequence and analysis of a broad-host range lytic bacteriophage that infects the Bacillus cereus group. Virol. J. 2013, 10, 48. [Google Scholar] [CrossRef]
- Merabishvili, M.; Vandenheuvel, D.; Kropinski, A.M.; Mast, J.; De Vos, D.; Verbeken, G.; Noben, J.P.; Lavigne, R.; Vaneechoutte, M.; Pirnay, J.P. Characterization of newly isolated lytic bacteriophages active against Acinetobacter baumannii. PLoS ONE 2014, 9, e104853. [Google Scholar] [CrossRef]
- Brok-Volchanskaya, V.S.; Kadyrov, F.A.; Sivogrivov, D.E.; Kolosov, P.M.; Sokolov, A.S.; Shlyapnikov, M.G.; Kryukov, V.M.; Granovsky, I.E. Phage T4 SegB protein is a homing endonuclease required for the preferred inheritance of T4 tRNA gene region occurring in co-infection with a related phage. Nucleic Acids Res. 2008, 36, 2094–2105. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| # Genomes w tRNA/Total Genomes | Family | Order | DNA/RNA | Hosts | Length (Kb) | # tRNAs | Avg GC% |
|---|---|---|---|---|---|---|---|
| 1/24 | Dicistroviridae | Picornavirales | ssRNA(+) | Invertebrates | 9 | 1 | 45.31 |
| 1/9 | Lipothrixviridae | Ligamenvirales | dsDNA | Archaea | 32 | 1 | 35.66 |
| 1/47 | Luteoviridae | Unassigned | ssRNA(+) | Plants | 6 | 1 | 50.70 |
| 1/7 | Marseilleviridae | Unassigned | dsDNA | Amoeba | 372 | 2 | 44.19 |
| 1/6 | Nudiviridae | Unassigned | dsDNA | Insects and marine crustaceans | 145 | 1 | 25.53 |
| 1/97 | Polyomaviridae | Unassigned | dsDNA | Mammals and birds | 5 | 1 | 52.35 |
| 1/69 | Virgaviridae | Unassigned | ssRNA(+) | Plants | 11 | 1 | 48.53 |
| 2/48 | Poxviridae | “Megavirales” | dsDNA | Humans, vertebrates and arthropods | ~140 | 1 | 51.65 |
| 2/8 | Polydnaviridae | Unassigned | dsDNA | Parasitoid wasps | 185–564 | 7–8 | 33.72 |
| 3/6 | Ascoviridae | “Megavirales” | dsDNA | Insects | 173–198 | 1–3 | 42.67 |
| 3/47 | Inoviridae | Unassigned | ssDNA | Bacteria | ~5 | 1 | 44.30 |
| 3/67 | Retroviridae | Unassigned | ssRNA(+) | Vertebrates | 6–8 | 1 | 48.80 |
| 4/21 | Iridoviridae | Unassigned | dsDNA | Amphibia, fish and invertebrates | 123–190 | 1 | 39.92 |
| 5/11 | Fuselloviridae | Unassigned | dsDNA | Thermophilic archaea | ~16 | 1 | 38.37 |
| 5/6 | Mimiviridae | “Megavirales” | dsDNA | Amoeba | 600–1200 | 2–15 | 26.12 |
| 6/78 | Herpesviridae | Herpesvirales | dsDNA | Vertebrates | 119–203 | 1–18 | 62.38 |
| 7/97 | Adenoviridae | Unassigned | dsDNA | Vertebrates | 33–46 | 1 | 52.50 |
| 9/84 | Baculoviridae | Unassigned | dsDNA | Arthropods and crustacean | 81–178 | 1 | 44.01 |
| 21/24 | Phycodnaviridae | Unassigned | dsDNA | Alga | 170–469 | 2–14 | 39.77 |
| 115/584 | Podoviridae | Caudovirales | dsDNA | Archaea and Bacteria | 36–145 | 1–23 | 44.78 |
| 620/1981 | Siphoviridae | Caudovirales | dsDNA | Archaea and Bacteria | 14–280 | 1–43 | 55.51 |
| 776/1079 | Myoviridae | Caudovirales | dsDNA | Archaea and Bacteria | 32–497 | 1–36 | 41.59 |
| # Genomes | Genus | Family | Phylum | Domain |
|---|---|---|---|---|
| 55 | Mycobacterium | Mycobacteriaceae | Actinobacteria | Bacteria |
| 9 | Streptomyces | Streptomycetaceae | Actinobacteria | Bacteria |
| 1 | Gordonia | Gordoniaceae | Actinobacteria | Bacteria |
| 7 | Cellulophaga | Flavobacteriaceae | Bacteroidetes | Bacteria |
| 2 | Synechococcus | Synechococcaceae | Cyanobacteria | Bacteria |
| 1 | Halogranum | Haloferacaceae | Euryarchaeota | Archaea |
| 13 | Bacillus | Bacillaceae | Firmicutes | Bacteria |
| 1 | Enterococcus | Enterobacteriaceae | Firmicutes | Bacteria |
| 1 | Staphylococcus | Staphylococcaceae | Firmicutes | Bacteria |
| 1 | Lactobacillus | Lactobacillaceae | Firmicutes | Bacteria |
| 10 | Listeria | Listeriaceae | Firmicutes | Bacteria |
| 5 | Acinetobacter | Moraxellaceae | Proteobacteria | Bacteria |
| 14 | Aeromonas | Aeromonadaceae | Proteobacteria | Bacteria |
| 12 | Caulobacter | Caulobacteraceae | Proteobacteria | Bacteria |
| 4 | Citrobacter | Enterobacteriaceae | Proteobacteria | Bacteria |
| 5 | Cronobacter | Enterobacteriaceae | Proteobacteria | Bacteria |
| 3 | Erwinia | Enterococcaceae | Proteobacteria | Bacteria |
| 23 | Escherichia | Enterobacteriaceae | Proteobacteria | Bacteria |
| 7 | Klebsiella | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Pectobacterium | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Providencia | Enterobacteriaceae | Proteobacteria | Bacteria |
| 7 | Pseudomonas | Pseudomonadaceae | Proteobacteria | Bacteria |
| 1 | Roseobacter | Rhodobacteraceae | Proteobacteria | Bacteria |
| 17 | Salmonella | Enterobacteriaceae | Proteobacteria | Bacteria |
| 4 | Shigella | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Sphingobium | Sphingomonadaceae | Proteobacteria | Bacteria |
| 3 | Stenotrophomonas | Xanthomonadaceae | Proteobacteria | Bacteria |
| 9 | Vibrio | Vibrionaceae | Proteobacteria | Bacteria |
| 1 | Yersinia | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Agrobacterium | Rhizobiaceae | Proteobacteria | Bacteria |
| 1 | Enterobacter | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Pseudoalteromonas | Pseudoalteromonadaceae | Proteobacteria | Bacteria |
| 1 | Ralstonia | Burkholderiaceae | Proteobacteria | Bacteria |
| 3 | Serratia | Enterobacteriaceae | Proteobacteria | Bacteria |
| 1 | Sulfitobacter | Rhodobacteraceae | Proteobacteria | Bacteria |
| 1 | Cafeteria | Cafeteriaceae | Stramenopiles | Eukarya |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgado, S.; Vicente, A.C. Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses 2019, 11, 180. https://doi.org/10.3390/v11020180
Morgado S, Vicente AC. Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses. 2019; 11(2):180. https://doi.org/10.3390/v11020180
Chicago/Turabian StyleMorgado, Sergio, and Ana Carolina Vicente. 2019. "Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes" Viruses 11, no. 2: 180. https://doi.org/10.3390/v11020180
APA StyleMorgado, S., & Vicente, A. C. (2019). Global In-Silico Scenario of tRNA Genes and Their Organization in Virus Genomes. Viruses, 11(2), 180. https://doi.org/10.3390/v11020180