BK Polyomavirus MicroRNA Levels in Exosomes Are Modulated by Non-Coding Control Region Activity and Down-Regulate Viral Replication When Delivered to Non-Infected Cells Prior to Infection

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Urine Sampling
2.2. Cell Cultures and BkPyV Molecular Clones
2.3. BKPyV Infection of Cos-7 and RPTECs
2.4. Plasmid Exosomes BKPyV miRNA Expression in Cos-7 Cells
2.5. Exosome Enriched Vesicles Extraction
2.6. Exosome Addition and Anti BKPyV-miRNA Inhibition in Cos-7 Cells
2.7. BKPyV DNA Quantification
2.8. BKPyV Pre-miRNA and Mature miRNA Quantification
2.9. siRNA knock-Down of Sp1 and Immunoblotting
2.10. NCCR Sequencing
2.11. Statistical Tests
3. Results
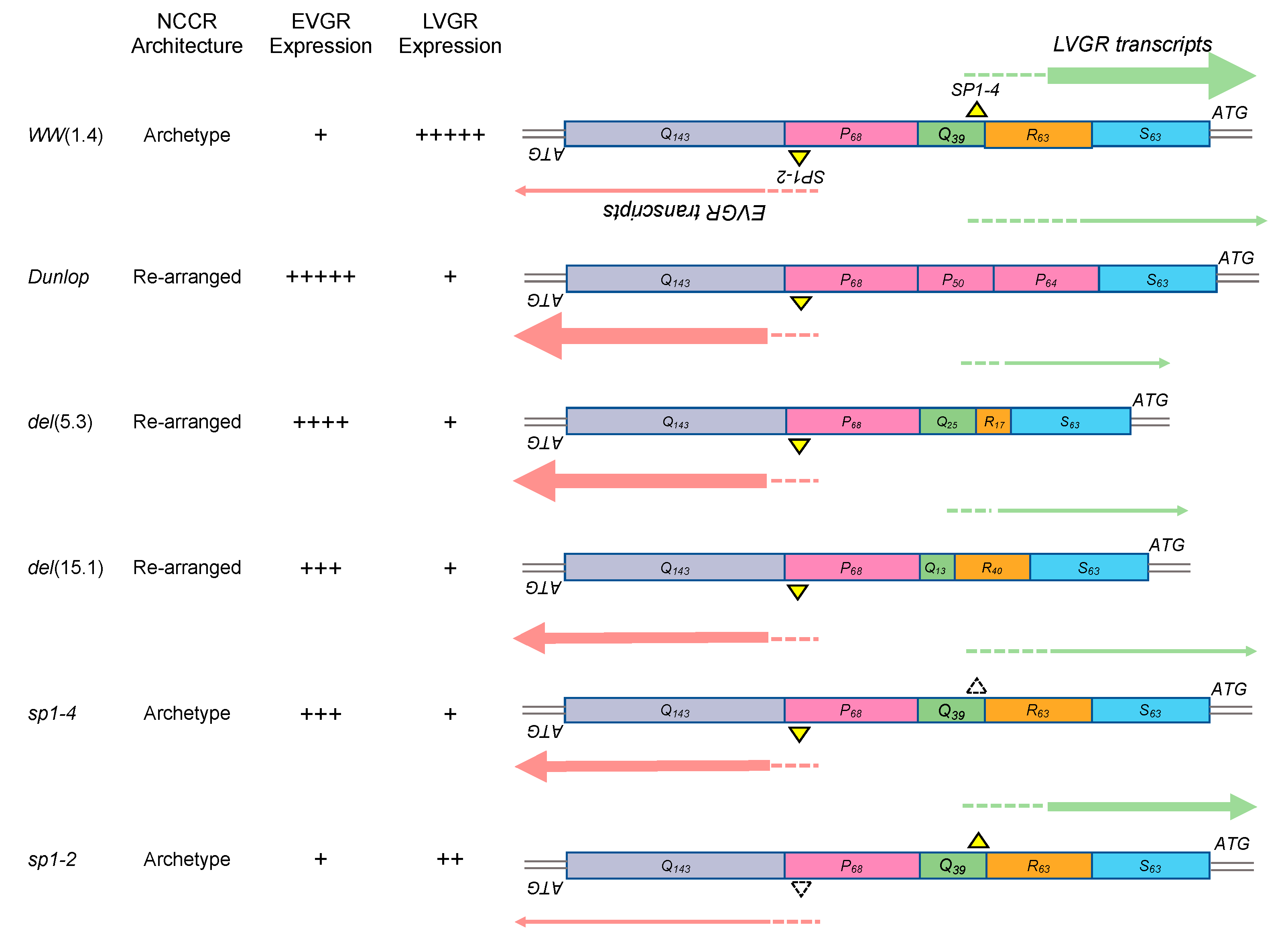
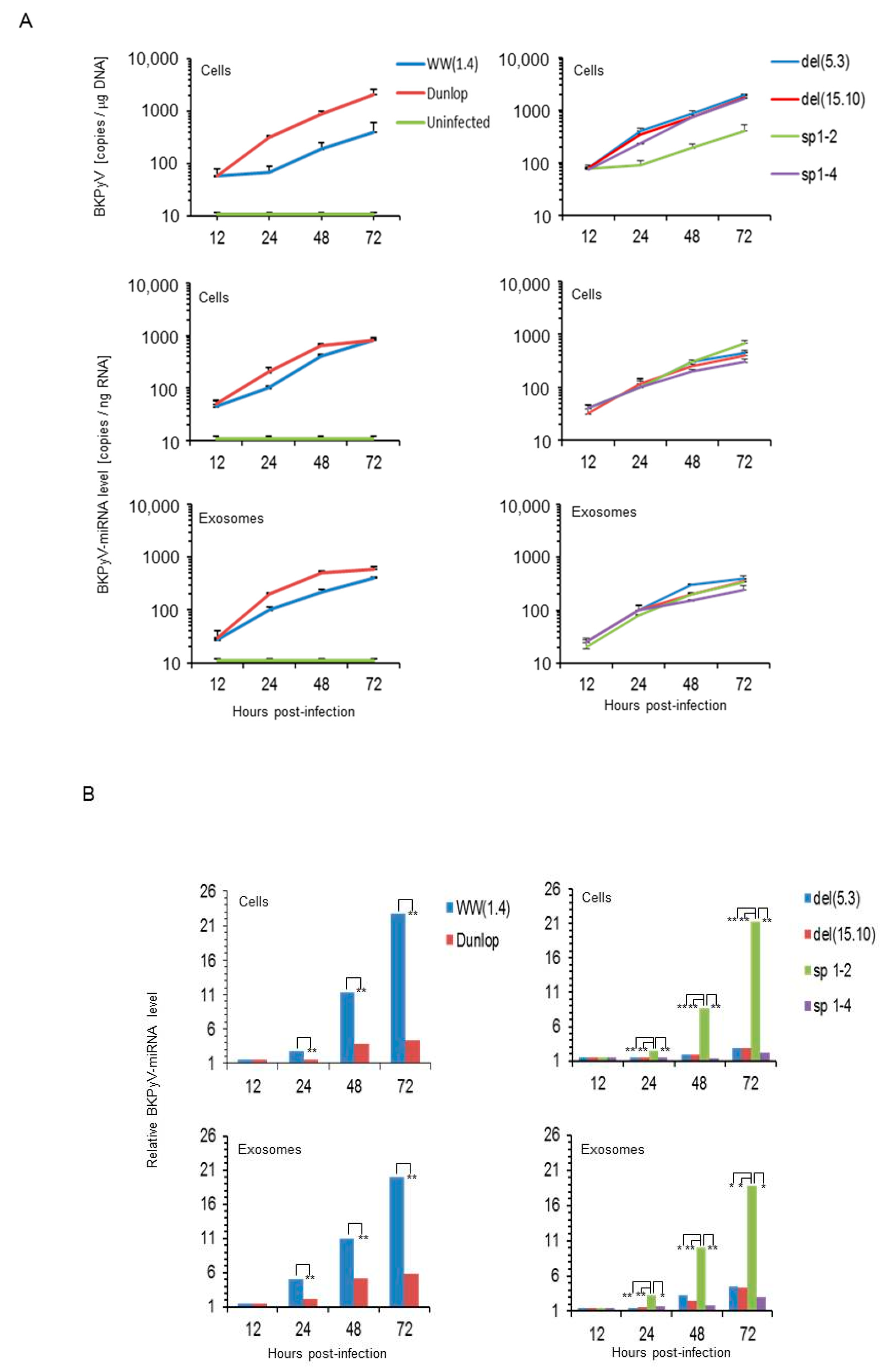
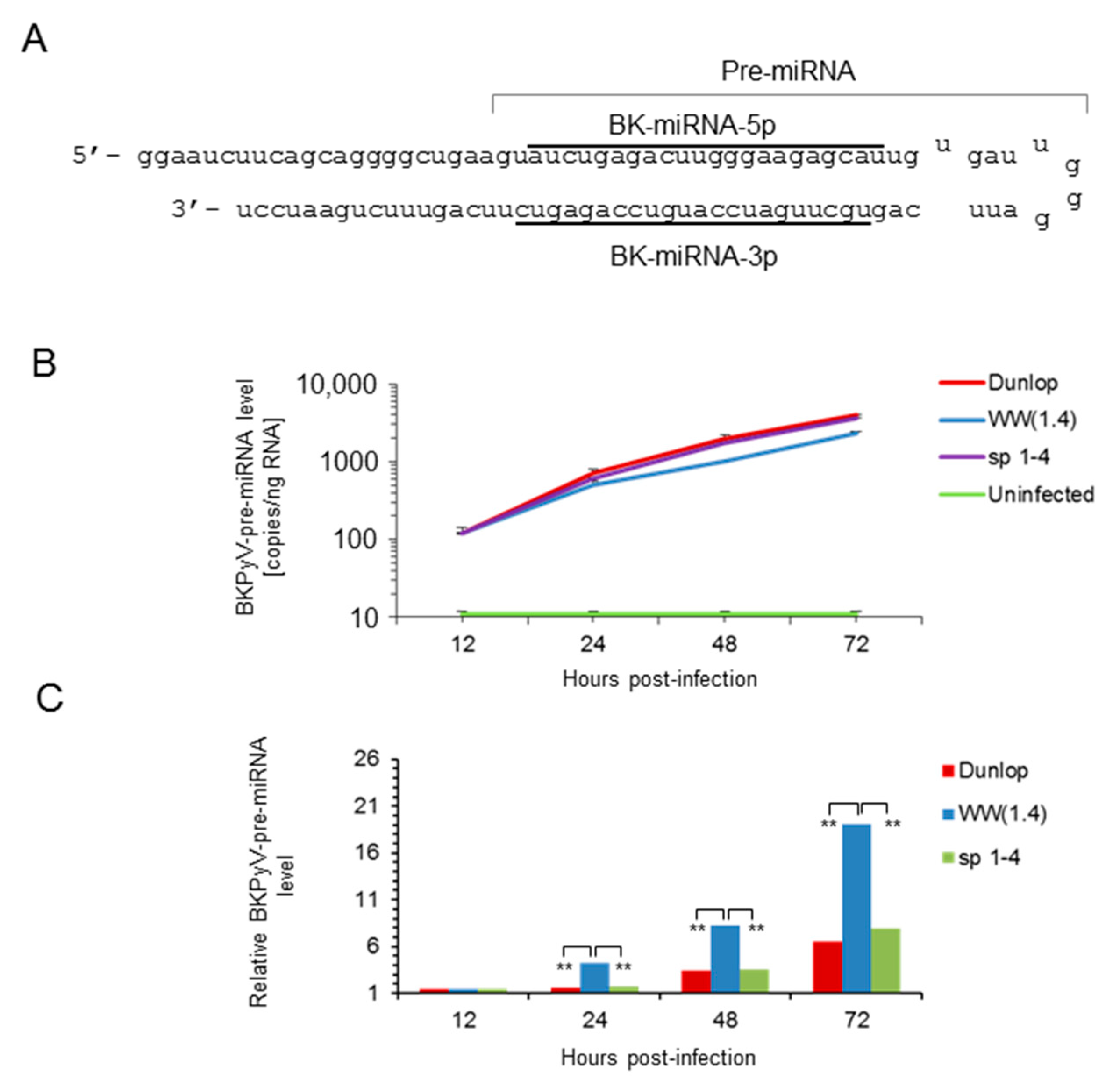
3.1. Rearranged NCCR-BKPyV Variants Show Higher Viral Loads But 10-Fold Lower miRNA Expression than Archetype NCCR BKPyV
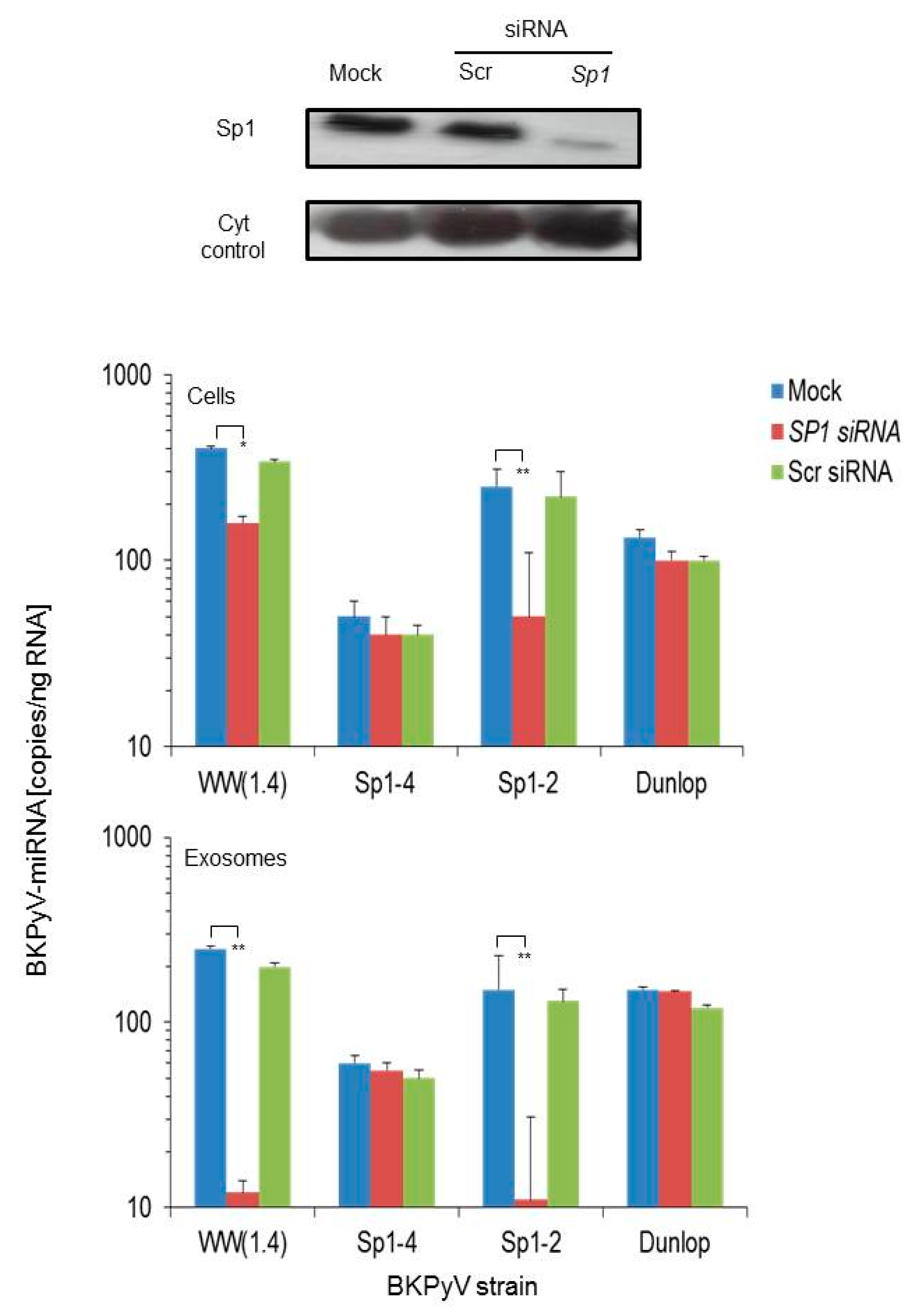
3.2. Sp1 Expression Levels Modulate BKPyV miRNA Levels in Archetype NCCR Virus And Variants With Low EVGR Expression But Not in Viral Variants Having an Activated EVGR Expression
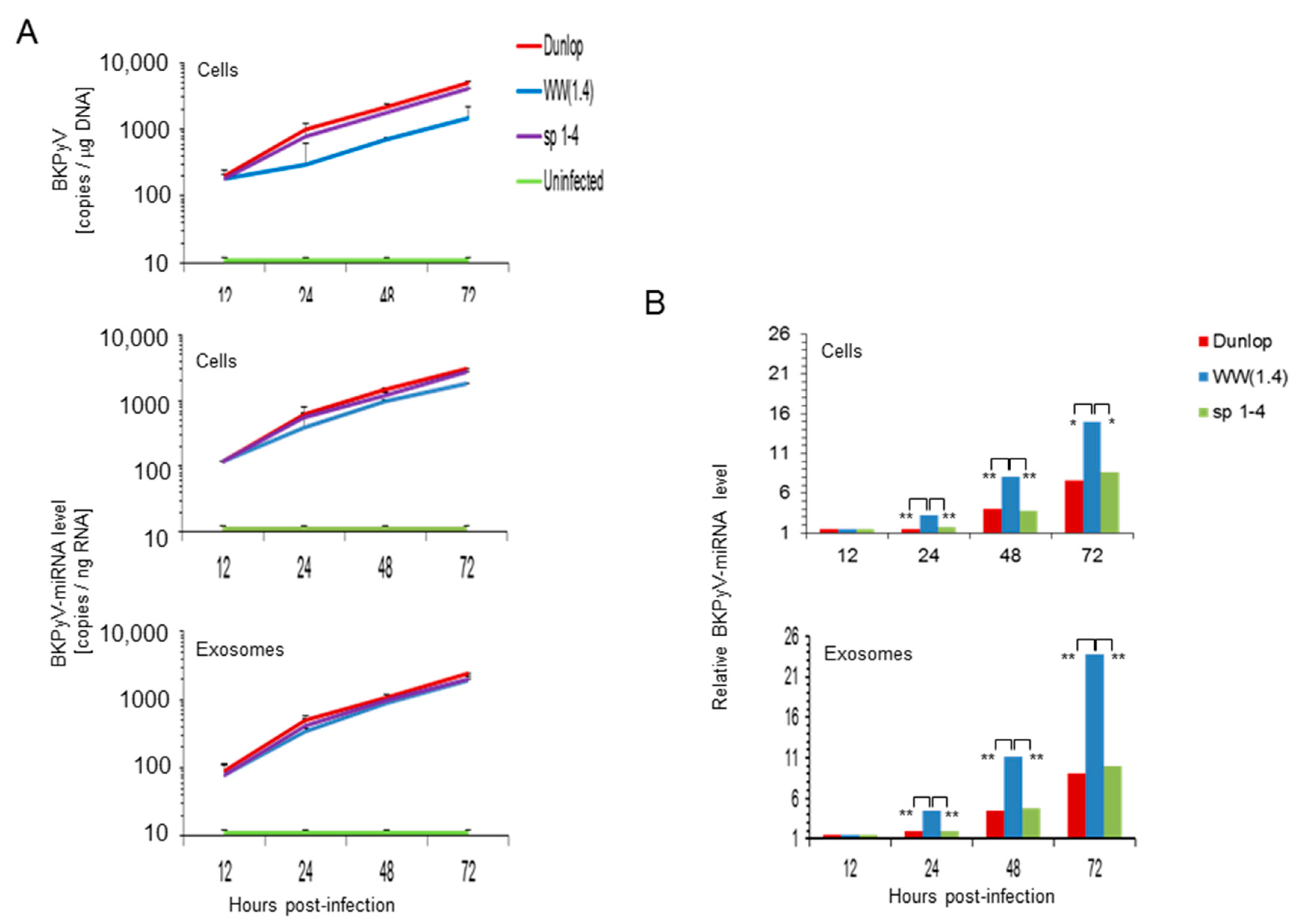
3.3. BKPyV miRNA Levels in Primary Human Renal Tubular Epithelial Cells (RPTECs) Inversely Correlate with the Evgr-Activity and Replication Rate of Archetype and rr-NCCR-Viruses
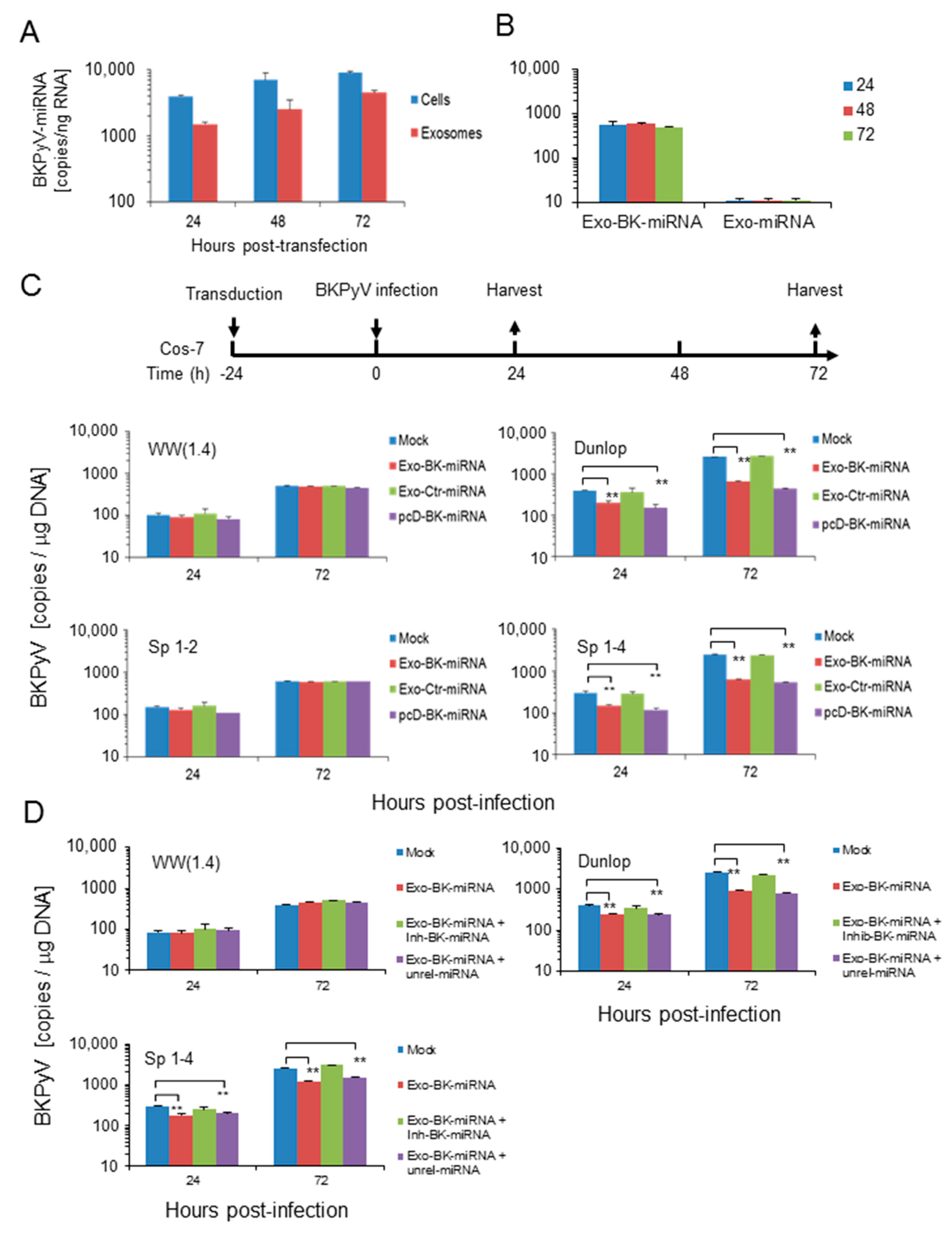
3.4. Pre-Infection Addition of BKPyV-Exosome Preparations Containing BKPyV miRNA Inhibit BKPyV Replication of Dunlop or sp1-4 Mutant But Have No Effect On Archetype BKPyV or sp1-2 Mutant Strains
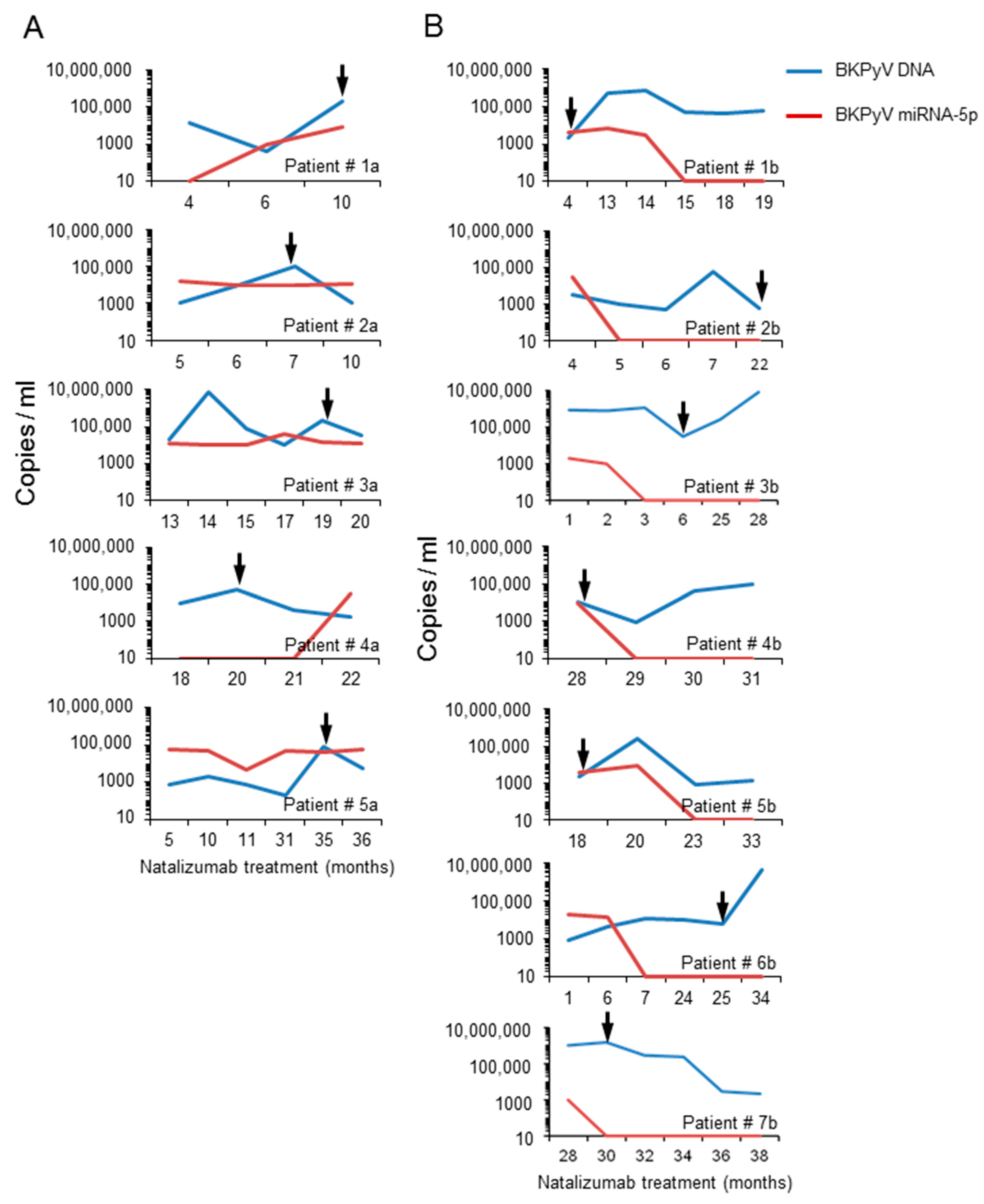
3.5. Analysis of the BKPyV NCCR Architecture and Exosomes Content in Urine Samples of Immunocompromised Patients
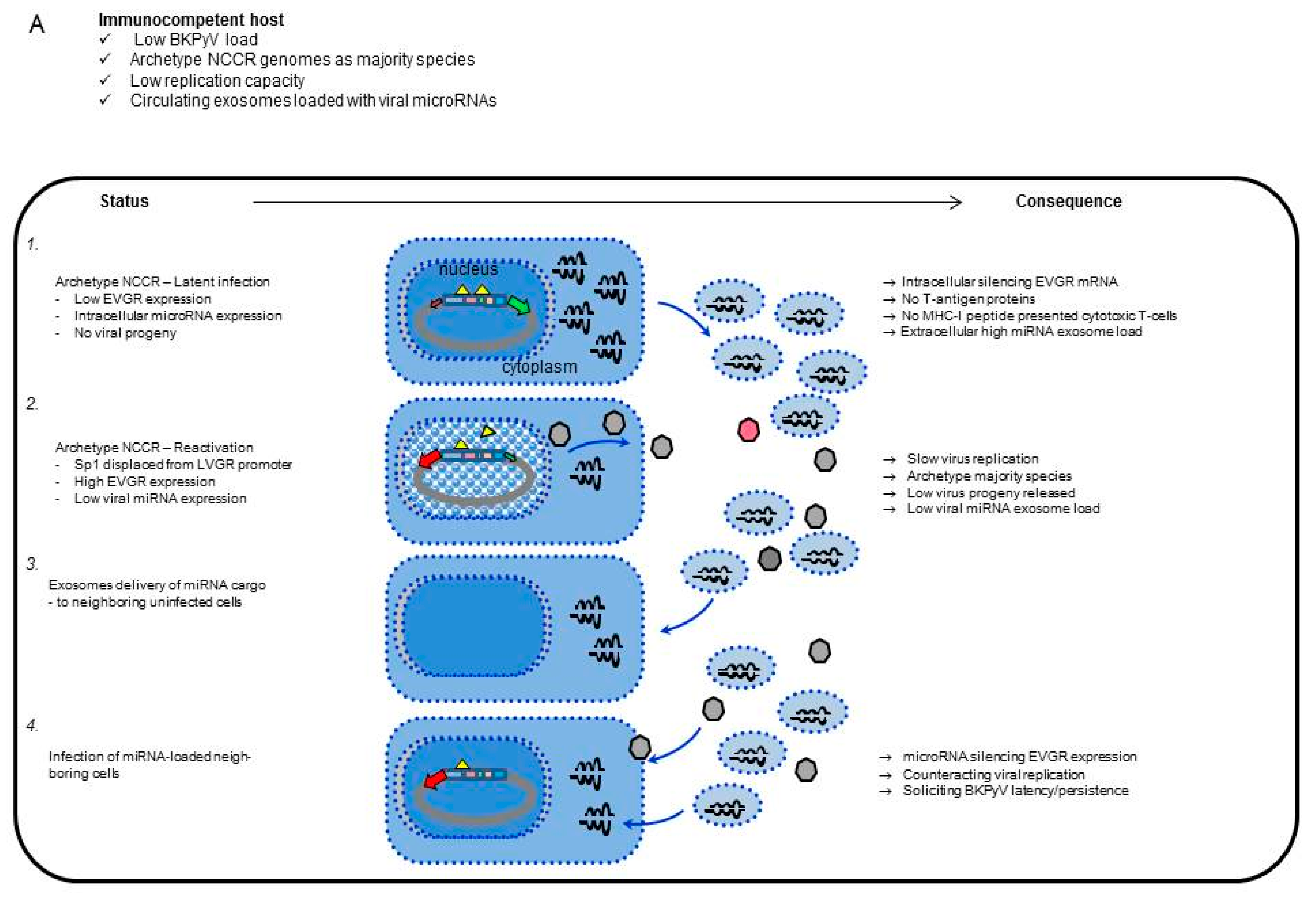
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- DeCaprio, J.A.; Imperiale, M.J.; Major, E.O. Polyomaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; LWW: Philadelphia, PA, USA, 2013; pp. 1633–1661. [Google Scholar]
- Greenlee, J.E.; Hirsch, H.H. Polyomaviruses. In Clinical Virology, 4th ed.; Microbiology, A.A.S.F., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2017; pp. 599–623. [Google Scholar]
- Rinaldo, C.H.; Hirsch, H.H. The human polyomaviruses: From orphans and mutants to patchwork family. APMIS 2013, 121, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Babel, N.; Comoli, P.; Friman, V.; Ginevri, F.; Jardine, A.; Lautenschlager, I.; Legendre, C.; Midtvedt, K.; Munoz, P.; et al. European perspective on human polyomavirus infection, replication and disease in solid organ transplantation. Clin. Microbiol. Infect. 2014, 20 (Suppl. 7), 74–88. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, S.; Dalianis, T.; Hanssen Rinaldo, C.; Koskenvuo, M.; Pegoraro, A.; Einsele, H.; Cordonnier, C.; Hirsch, H.H.; Group, E. ECIL guidelines for the prevention, diagnosis and treatment of BK polyomavirus-associated haemorrhagic cystitis in haematopoietic stem cell transplant recipients. J. Antimicrob. Chemother. 2018, 73, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Knowles, W.A. Discovery and epidemiology of the human polyomaviruses BK virus (BKV) and JC virus (JCV). Adv. Exp. Med. Biol. 2006, 577, 19–45. [Google Scholar] [PubMed]
- Hirsch, H.H.; Steiger, J. Polyomavirus BK. Lancet Infect. Dis. 2003, 3, 611–623. [Google Scholar] [CrossRef]
- Imperiale, M.J.; Jiang, M. Polyomavirus Persistence. Annu. Rev. Virol. 2016, 3, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Kean, J.M.; Rao, S.; Wang, M.; Garcea, R.L. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009, 5, e1000363. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, D.; Andresen, P.A.; Gosert, R.; Binggeli, S.; Strom, E.H.; Bodaghi, S.; Rinaldo, C.H.; Hirsch, H.H. Human polyomavirus type 1 (BK virus) agnoprotein is abundantly expressed but immunologically ignored. Clin. Vaccine Immunol. 2007, 14, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, S.; Comoli, P.; Boesch, R.; Azzi, A.; Gosert, R.; Leuenberger, D.; Ginevri, F.; Hirsch, H.H. Antibody Responses to Recombinant Polyomavirus BK Large T and VP1 Proteins in Pediatric Kidney Transplant Patients. J. Clin. Micro 2009, 47, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Brennan, D.C.; Cuburu, N.; Storch, G.A.; Viscidi, R.P.; Randhawa, P.S.; Buck, C.B. Neutralization serotyping of BK polyomavirus infection in kidney transplant recipients. PLoS Pathog. 2012, 8, e1002650. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Adam, C.; Hirsch, H.H.; Janssen, M.W.; Wolf, M.; Dirks, J.; Kardas, P.; Ahlenstiel-Grunow, T.; Pape, L.; Rohrer, T.; et al. BK Polyomavirus-Specific Cellular Immune Responses Are Age-Dependent and Strongly Correlate With Phases of Virus Replication. Am. J. Transplant. 2014, 14, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Funk, G.A.; Gosert, R.; Comoli, P.; Ginevri, F.; Hirsch, H.H. Polyomavirus BK replication dynamics in vivo and in silico to predict cytopathology and viral clearance in kidney transplants. Am. J. Transplant. 2008, 8, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.; Vats, A.; Shapiro, R. Monitoring for polyomavirus BK and JC in urine: Comparison of quantitative polymerase chain reaction with urine cytology. Transplantation 2005, 79, 984–986. [Google Scholar] [CrossRef] [PubMed]
- Drachenberg, C.B.; Papadimitriou, J.C.; Hirsch, H.H.; Wali, R.; Crowder, C.; Nogueira, J.; Cangro, C.B.; Mendley, S.; Mian, A.; Ramos, E. Histological patterns of polyomavirus nephropathy: Correlation with graft outcome and viral load. Am. J. Transplant. 2004, 4, 2082–2092. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Knowles, W.; Dickenmann, M.; Passweg, J.; Klimkait, T.; Mihatsch, M.J.; Steiger, J. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N. Engl. J. Med. 2002, 347, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, P.; Uhrmacher, J.; Pasculle, W.; Vats, A.; Shapiro, R.; Eghtsead, B.; Weck, K. A comparative study of BK and JC virus infections in organ transplant recipients. J. Med. Virol. 2005, 77, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Abend, J.R.; Johnson, S.F.; Imperiale, M.J. The role of polyomaviruses in human disease. Virology 2009, 384, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Helmersen, D.S.; Taub, K.; Hirsch, H.H.; Johnson, A. Renal Failure Five Years After Lung Transplantation Due to Polyomavirus BK-Associated Nephropathy. Am. J. Transplant. 2010, 10, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, M.J. Polyomavirus miRNAs: The beginning. Curr. Opin. Virol. 2014, 7, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ajuh, E.; Wu, Z.; Kraus, E.; Weissbach, F.H.; Bethge, T.; Gosert, R.; Fischer, N.; Hirsch, H.H. Novel Human Polyomavirus non-coding control regions differ in bi-directional gene expression according to host cell, large T-antigen expression and clinically occurring rearrangements. J. Virol. 2018, 92, e02231-17. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Rinaldo, C.H.; Funk, G.A.; Egli, A.; Ramos, E.; Drachenberg, C.B.; Hirsch, H.H. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 2008, 205, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.H.; Hirsch, H.H.; Rinaldo, C.H. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J. Med. Virol. 2009, 81, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.M.; Gupta, G.; Vats, A.; Shapiro, R.; Randhawa, P.S. Polyomavirus BK non-coding control region rearrangements in health and disease. J. Med. Virol. 2007, 79, 1199–11207. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kardas, P.; Major, E.O.; Hirsch, H.H. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J. Virol. 2010, 84, 10448–10456. [Google Scholar] [CrossRef] [PubMed]
- Khanna, N.; Wolbers, M.; Mueller, N.J.; Garzoni, C.; Du Pasquier, R.A.; Fux, C.A.; Vernazza, P.; Bernasconi, E.; Viscidi, R.; Battegay, M.; et al. JC virus-specific immune responses in human immunodeficiency virus type 1 patients with progressive multifocal leukoencephalopathy. J. Virol. 2009, 83, 4404–4411. [Google Scholar] [CrossRef] [PubMed]
- Bethge, T.; Hachemi, H.A.; Manzetti, J.; Gosert, R.; Schaffner, W.; Hirsch, H.H. Sp1 sites in the noncoding control region of BK polyomavirus are key regulators of bidirectional viral early and late gene expression. J. Virol. 2015, 89, 3396–33411. [Google Scholar] [CrossRef] [PubMed]
- Bethge, T.; Ajuh, E.; Hirsch, H.H. Imperfect Symmetry of Sp1 and Core Promoter Sequences Regulates Early and Late Virus Gene Expression of the Bidirectional BK Polyomavirus Noncoding Control Region. J. Virol. 2016, 90, 10083–10101. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Grundhoff, A.; Tevethia, S.; Treisman, R.; Pipas, J.M.; Ganem, D. Expression and function of microRNAs in viruses great and small. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Lagatie, O.; Tritsmans, L.; Stuyver, L.J. The miRNA world of polyomaviruses. Virol. J. 2013, 10, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Fink, L.H.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily conserved function of a viral microRNA. J. Virol. 2008, 82, 9823–9828. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Grundhoff, A.T.; Tevethia, S.; Pipas, J.M.; Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 2005, 435, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, P.; Doering, A.; Sullivan, C.S.; Pal, A.; Peden, K.W.; Lewis, A.M.; Pipas, J.M. Complete nucleotide sequence of polyomavirus SA12. J. Virol. 2005, 79, 13094–13104. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and microRNAs. Nat. Genet. 2006, 38, 25S–30S. [Google Scholar] [CrossRef] [PubMed]
- Bauman, Y.; Mandelboim, O. MicroRNA based immunoevasion mechanism of human polyomaviruses. RNA Biol. 2011, 8, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Cioni, M.; Leboeuf, C.; Comoli, P.; Ginevri, F.; Hirsch, H.H. Characterization of Immunodominant BK Polyomavirus 9mer Epitope T Cell Responses. Am. J. Transplant. 2016, 16, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Leboeuf, C.; Wilk, S.; Achermann, R.; Binet, I.; Golshayan, D.; Hadaya, K.; Hirzel, C.; Hoffmann, M.; Huynh-Do, U.; Koller, M.T.; et al. BK Polyomavirus-Specific 9mer CD8 T Cell Responses Correlate With Clearance of BK Viremia in Kidney Transplant Recipients: First Report From the Swiss Transplant Cohort Study. Am. J. Transplant. 2017, 17, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Safak, M.; Khalili, K. Regulation of gene expression in primate polyomaviruses. J. Virol. 2009, 83, 10846–10856. [Google Scholar] [CrossRef] [PubMed]
- Broekema, N.M.; Imperiale, M.J. miRNA regulation of BK polyomavirus replication during early infection. Proc. Natl. Acad. Sci. USA 2013, 110, 8200–8205. [Google Scholar] [CrossRef] [PubMed]
- Martelli, F.; Giannecchini, S. Polyomavirus microRNAs circulating in biological fluids during viral persistence. Rev. Med. Virol. 2017, e1927. [Google Scholar] [CrossRef] [PubMed]
- Rocca, A.; Martelli, F.; Delbue, S.; Ferrante, P.; Bartolozzi, D.; Azzi, A.; Giannecchini, S. The JCPYV DNA load inversely correlates with the viral microrna expression in blood and cerebrospinal fluid of patients at risk of PML. J. Clin. Virol. 2015, 70, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Lee, Y.H.; Seo, J.W.; Moon, H.; Kim, J.S.; Kim, Y.G.; Jeong, K.H.; Moon, J.Y.; Lee, T.W.; Ihm, C.G.; et al. Urinary exosomal viral microRNA as a marker of BK virus nephropathy in kidney transplant recipients. PLoS ONE 2017, 12, e0190068. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, I.; Clausi, V.; Nukuzuma, S.; Della Malva, N.; Nosi, D.; Giannecchini, S. Polyomavirus JC microRNA expression after infection in vitro. Virus Res. 2016, 213, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, I.; Martelli, F.; Repice, A.; Massacesi, L.; Azzi, A.; Giannecchini, S. Detection of JCPyV microRNA in blood and urine samples of multiple sclerosis patients under natalizumab therapy. J. Neurovirol. 2015, 21, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Delbue, S.; Elia, F.; Carloni, C.; Pecchenini, V.; Franciotta, D.; Gastaldi, M.; Colombo, E.; Signorini, L.; Carluccio, S.; Bellizzi, A.; et al. JC virus urinary excretion and seroprevalence in natalizumab-treated multiple sclerosis patients. J. Neurovirol. 2015, 21, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Gluzman, Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell 1981, 23, 175–182. [Google Scholar] [CrossRef]
- Henriksen, S.; Mittelholzer, C.; Gosert, R.; Hirsch, H.H.; Rinaldo, C.H. Human BK Polyomavirus Plasmid pBKV (34-2) (Dunlop) Contains Mutations Not Found in the Originally Published Sequences. Genome Announc. 2015, 3, e00046-15. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Yakhontova, K.; Lu, M.; Manzetti, J. BK Polyomavirus Replication in Renal Tubular Epithelial Cells Is Inhibited by Sirolimus but Activated by Tacrolimus Through a Pathway Involving FKBP-12. Am. J. Transplant. 2016, 16, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sharma, B.N.; Linder, S.; Gutteberg, T.J.; Hirsch, H.H.; Rinaldo, C.H. Characteristics of polyomavirus BK (BKPyV) infection in primary human urothelial cells. Virology 2013, 440, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tremolada, S.; Delbue, S.; Castagnoli, L.; Allegrini, S.; Miglio, U.; Boldorini, R.; Elia, F.; Gordon, J.; Ferrante, P. Mutations in the external loops of BK virus VP1 and urine viral load in renal transplant recipients. J. Cell Physiol. 2010, 222, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Bernhoff, E.; Gutteberg, T.J.; Sandvik, K.; Hirsch, H.H.; Rinaldo, C.H. Cidofovir Inhibits Polyomavirus BK Replication in Human Renal Tubular Cells Downstream of Viral Early Gene Expression. Am. J. Transplant. 2008, 8, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Binet, I.; Nickeleit, V.; Hirsch, H.H.; Prince, O.; Dalquen, P.; Gudat, F.; Mihatsch, M.J.; Thiel, G. Polyomavirus disease under new immunosuppressive drugs: A cause of renal graft dysfunction and graft loss. Transplantation 1999, 67, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Steiger, J.; Mihatsch, M.J. Immunosuppression and BKV Nephropathy. N. Engl. J. Med. 2002, 347, 2079–2080. [Google Scholar]
- Zhao, L.; Imperiale, M.J. Identification of Rab18 as an Essential Host Factor for BK Polyomavirus Infection Using a Whole-Genome RNA Interference Screen. mSphere 2017, 2, e00291-174. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martelli, F.; Wu, Z.; Delbue, S.; Weissbach, F.H.; Giulioli, M.C.; Ferrante, P.; Hirsch, H.H.; Giannecchini, S. BK Polyomavirus MicroRNA Levels in Exosomes Are Modulated by Non-Coding Control Region Activity and Down-Regulate Viral Replication When Delivered to Non-Infected Cells Prior to Infection. Viruses 2018, 10, 466. https://doi.org/10.3390/v10090466
Martelli F, Wu Z, Delbue S, Weissbach FH, Giulioli MC, Ferrante P, Hirsch HH, Giannecchini S. BK Polyomavirus MicroRNA Levels in Exosomes Are Modulated by Non-Coding Control Region Activity and Down-Regulate Viral Replication When Delivered to Non-Infected Cells Prior to Infection. Viruses. 2018; 10(9):466. https://doi.org/10.3390/v10090466
Chicago/Turabian StyleMartelli, Francesco, Zongsong Wu, Serena Delbue, Fabian H. Weissbach, Maria Chiara Giulioli, Pasquale Ferrante, Hans H. Hirsch, and Simone Giannecchini. 2018. "BK Polyomavirus MicroRNA Levels in Exosomes Are Modulated by Non-Coding Control Region Activity and Down-Regulate Viral Replication When Delivered to Non-Infected Cells Prior to Infection" Viruses 10, no. 9: 466. https://doi.org/10.3390/v10090466
APA StyleMartelli, F., Wu, Z., Delbue, S., Weissbach, F. H., Giulioli, M. C., Ferrante, P., Hirsch, H. H., & Giannecchini, S. (2018). BK Polyomavirus MicroRNA Levels in Exosomes Are Modulated by Non-Coding Control Region Activity and Down-Regulate Viral Replication When Delivered to Non-Infected Cells Prior to Infection. Viruses, 10(9), 466. https://doi.org/10.3390/v10090466