Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses

 , and
, and 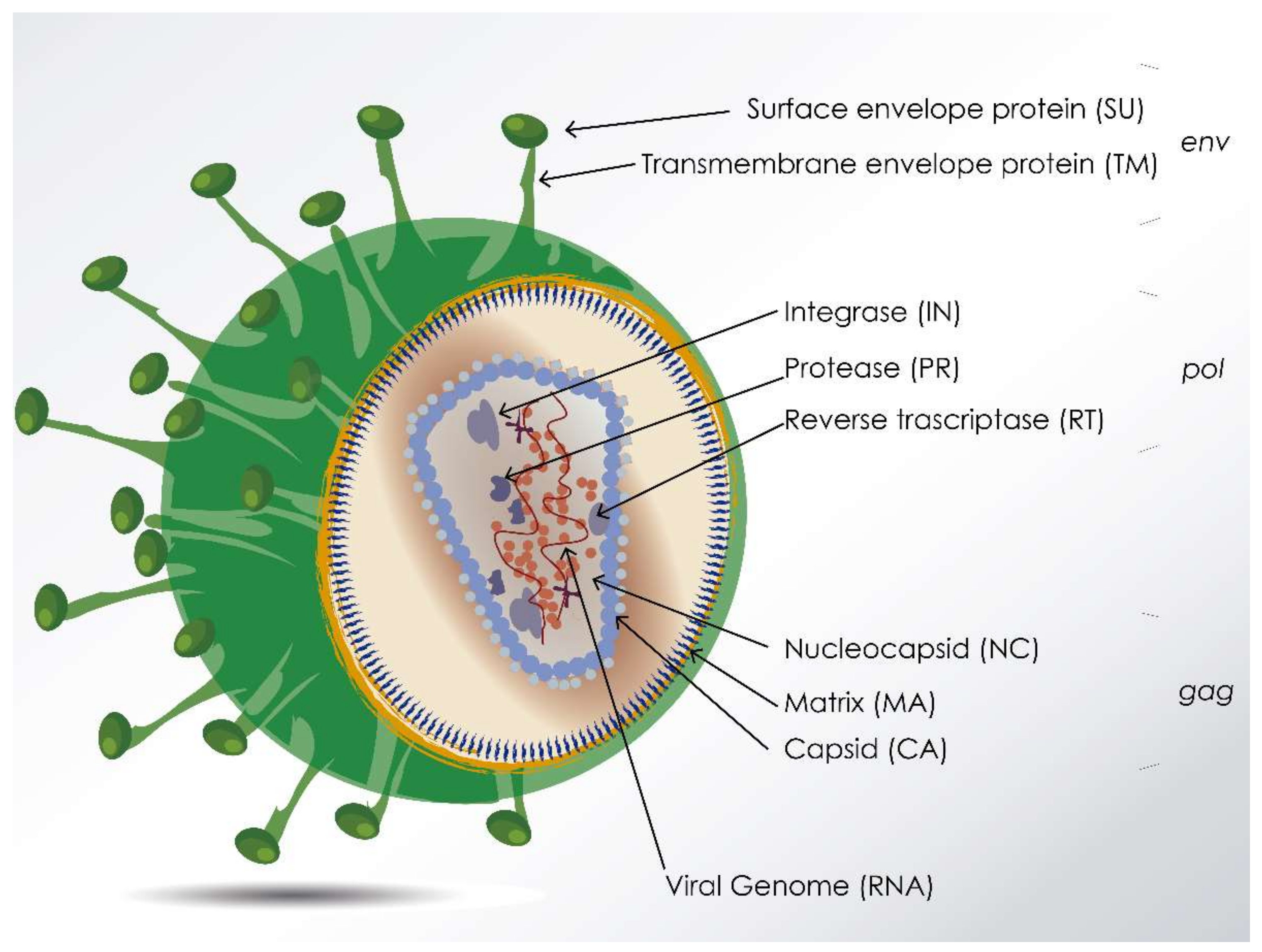{kind=link}
{kind=link}
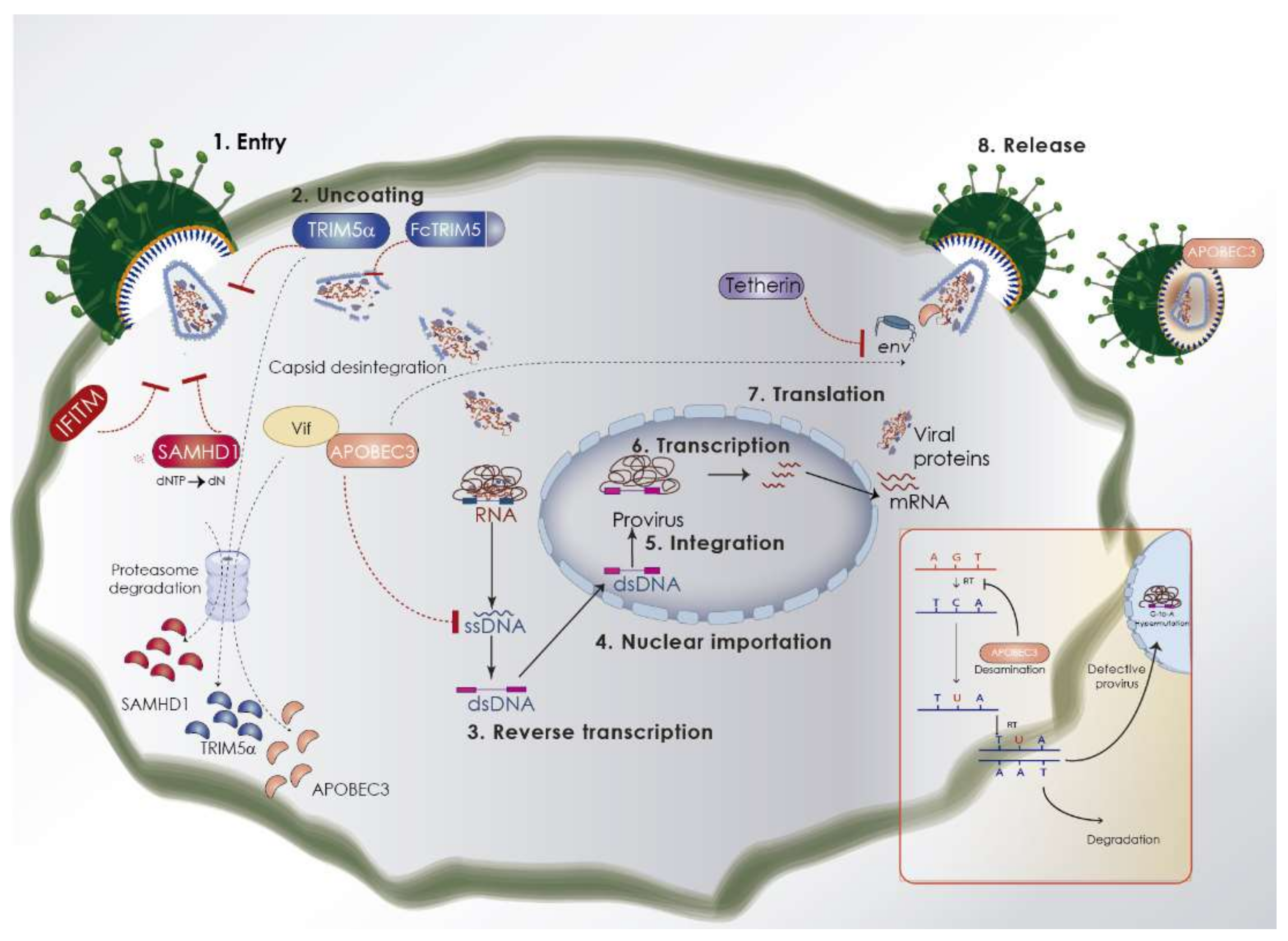{kind=link}
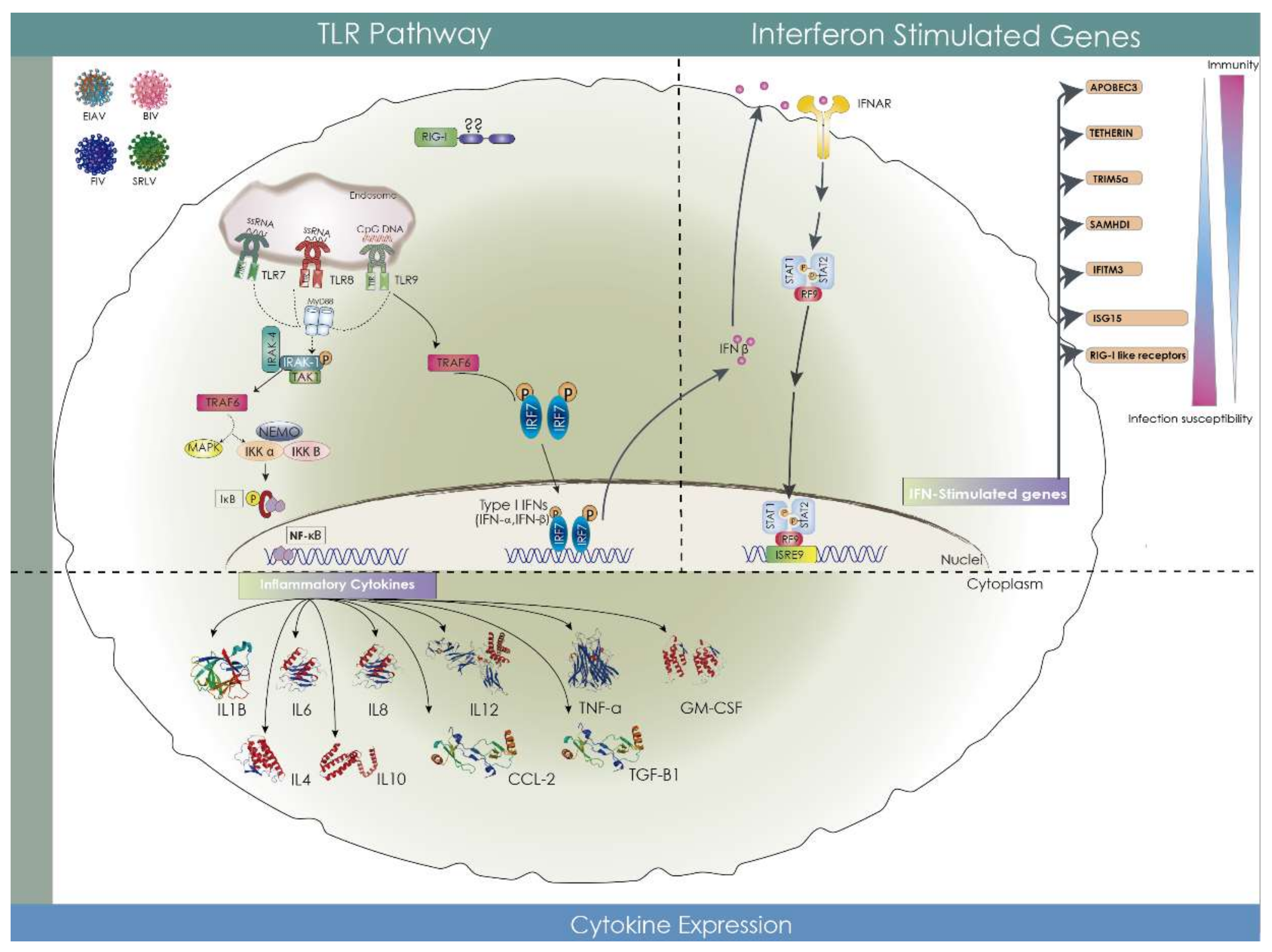{kind=link}
{kind=link}
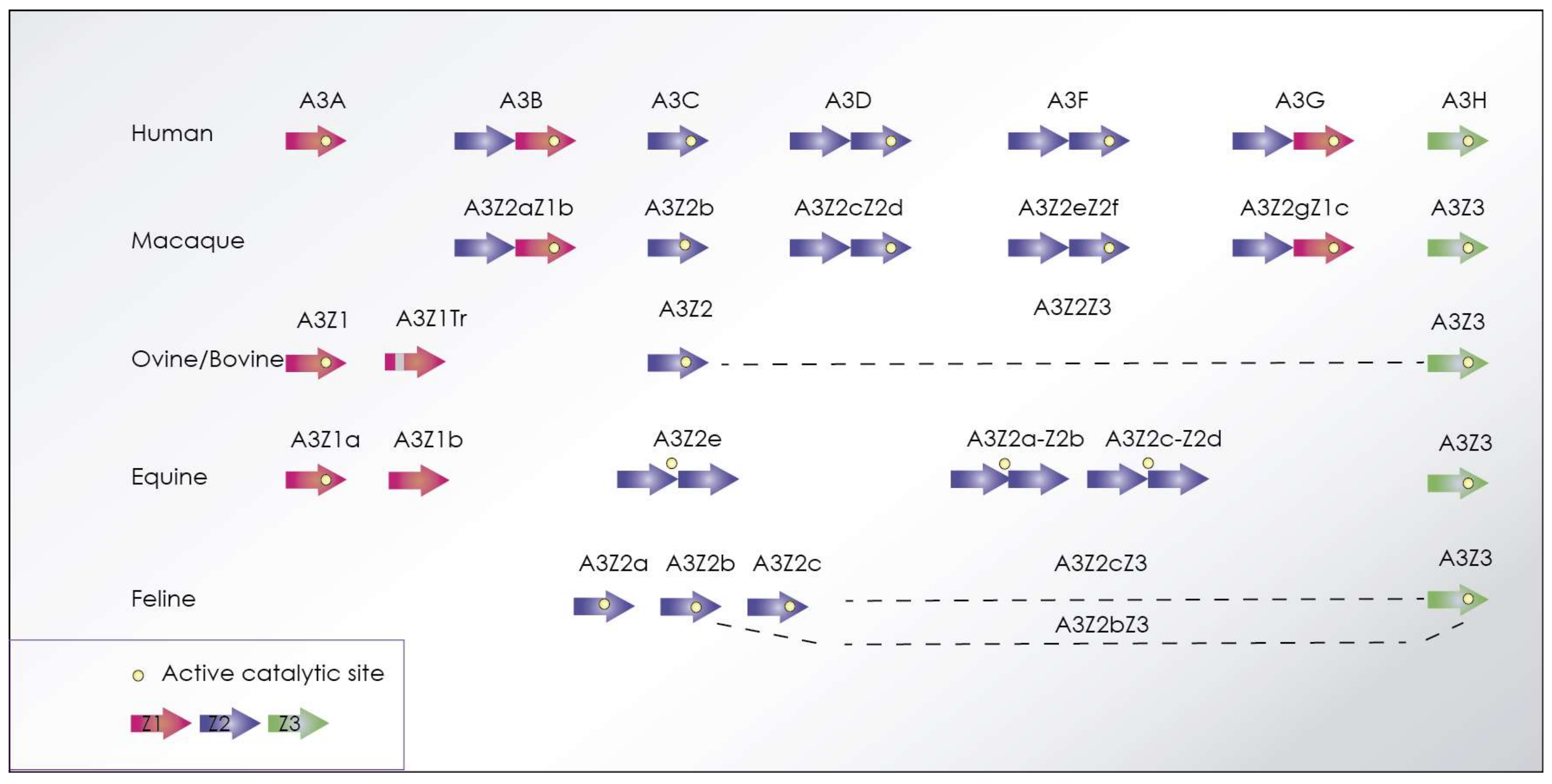{kind=link}
{kind=link}
Abstract
1. Introduction
2. Prevention and Control
2.1. Small Ruminant Lentiviruses (SRLV)
2.2. Equine Infectious Anemia Virus (EIAV)
2.3. Feline Immunodeficiency Virus (FIV)
2.4. Bovine Lentiviruses
3. Innate Immunity
3.1. SRLV
3.2. EIAV
3.3. FIV
3.4. BIV and JDV
4. TLR Signaling
4.1. SRLV
4.2. EIAV
4.3. FIV
4.4. BIV
5. Interferon
5.1. SRLV
5.2. EIAV
5.3. FIV
5.4. BIV
6. Host Restriction Factors
6.1. TRIM5α
6.1.1. SRLV
6.1.2. EIAV
6.1.3. FIV
6.1.4. BIV
6.2. APOBEC3
6.2.1. SRLV
6.2.2. EIAV
6.2.3. FIV
6.2.4. BIV
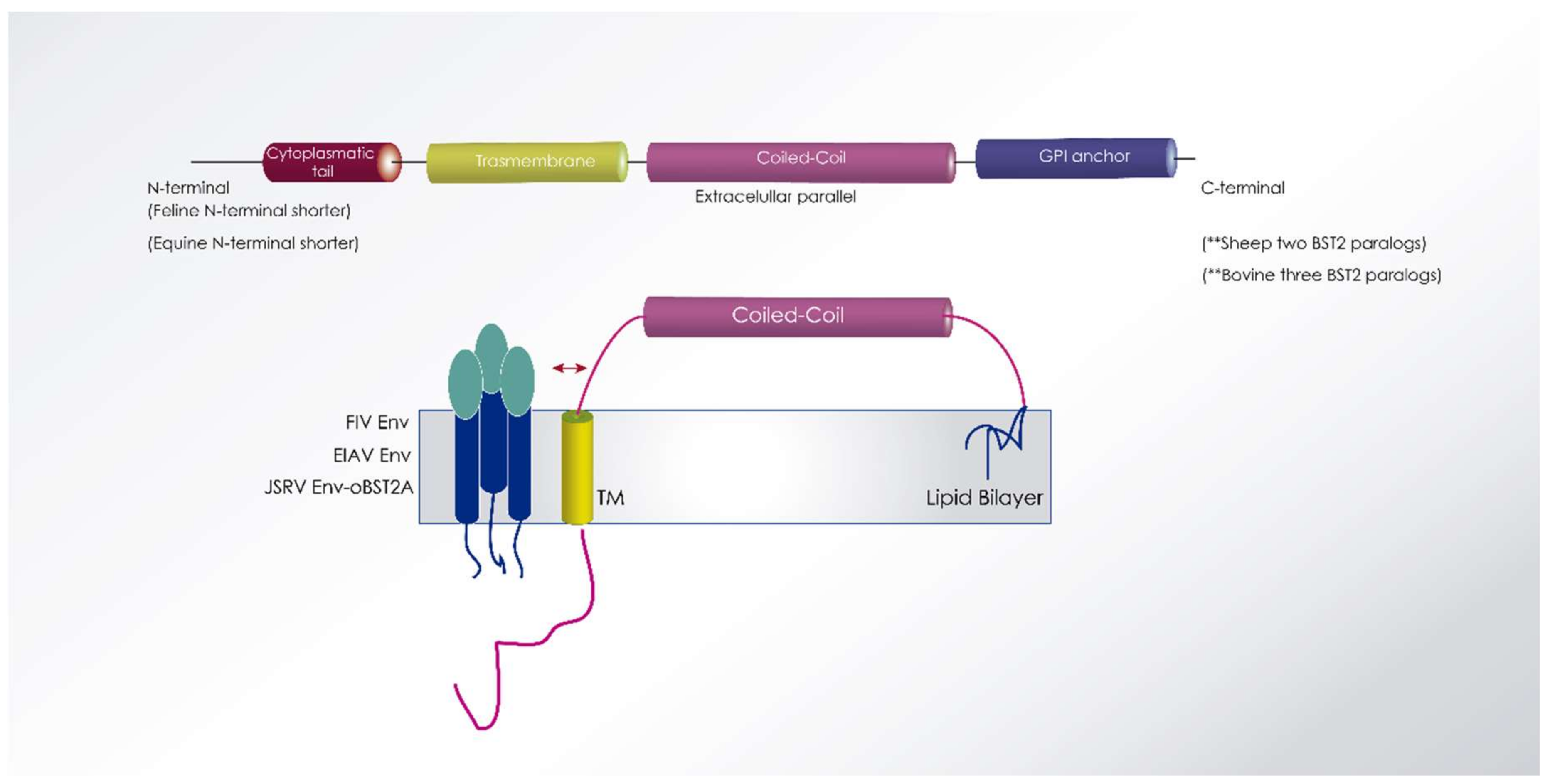
6.3. Tetherin
6.3.1. SRLV
6.3.2. EIAV
6.3.3. FIV
6.3.4. BIV
7. Other Host Protein Factors
7.1. SAMHD1
7.2. HEXIM1
7.3. Schlafen 11
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Minguijon, E.; Reina, R.; Perez, M.; Polledo, L.; Villoria, M.; Ramirez, H.; Leginagoikoa, I.; Badiola, J.J.; Garcia-Marin, J.F.; de Andres, D.; et al. Small ruminant lentivirus infections and diseases. Vet. Microbiol. 2015, 181, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.K.; Ezzelarab, C.; Cook, S.J.; Liu, C.; Horohov, D.; Issel, C.J.; Montelaro, R.C. Protective efficacy of centralized and polyvalent envelope immunogens in an attenuated equine lentivirus vaccine. PLoS Pathog. 2015, 11, e1004610. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J. Learning from the messengers: Innate sensing of viruses and cytokine regulation of immunity—Clues for treatments and vaccines. Viruses 2013, 5, 470–527. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Bloch, N.; Landau, N.R. Intrinsic host restrictions to HIV-1 and mechanisms of viral escape. Nat. Immunol. 2015, 16, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, R.G. Phylogenetic analysis of small ruminant lentiviruses. J. Gen. Virol. 1998, 79 Pt 8, 1951–1961. [Google Scholar] [CrossRef]
- Benavides, J.; Gomez, N.; Gelmetti, D.; Ferreras, M.C.; Garcia-Pariente, C.; Fuertes, M.; Garcia-Marin, J.F.; Perez, V. Diagnosis of the nervous form of Maedi-Visna infection with a high frequency in sheep in Castilla y Leon, Spain. Vet. Rec. 2006, 158, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Blacklaws, B.A. Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Gagnon, J.; Chebloune, Y. Comparative Analysis of Tat-Dependent and Tat-Deficient Natural Lentiviruses. Vet. Sci. 2015, 2, 293–348. [Google Scholar] [CrossRef] [PubMed]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, M.; Chatterji, U.; Schaffer, L.; de Rozieres, S.; Elder, J.H. Feline immunodeficiency virus OrfA alters gene expression of splicing factors and proteasome-ubiquitination proteins. Virology 2008, 371, 394–404. [Google Scholar] [CrossRef] [PubMed]
- De Andres, D.; Klein, D.; Watt, N.J.; Berriatua, E.; Torsteinsdottir, S.; Blacklaws, B.A.; Harkiss, G.D. Diagnostic tests for small ruminant lentiviruses. Vet. Microbiol. 2005, 107, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.P.; Clawson, M.L.; Chitko-Mckown, C.G.; Leymaster, K.A.; Smith, T.P.; Harhay, G.P.; White, S.N.; Herrmann-Hoesing, L.M.; Mousel, M.R.; Lewis, G.S.; et al. Reduced lentivirus susceptibility in sheep with TMEM154 mutations. PLoS Genet. 2012, 8, e1002467. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.; Carson, A.; Isaac, P. Genetic distinctiveness of the Herdwick sheep breed and two other locally adapted hill breeds of the UK. PLoS ONE 2014, 9, e87823. [Google Scholar] [CrossRef] [PubMed]
- White, S.N.; Mousel, M.R.; Herrmann-Hoesing, L.M.; Reynolds, J.O.; Leymaster, K.A.; Neibergs, H.L.; Lewis, G.S.; Knowles, D.P. Genome-wide association identifies multiple genomic regions associated with susceptibility to and control of ovine lentivirus. PLoS ONE 2012, 7, e47829. [Google Scholar] [CrossRef] [PubMed]
- White, S.N.; Mousel, M.R.; Reynolds, J.O.; Lewis, G.S.; Herrmann-Hoesing, L.M. Common promoter deletion is associated with 3.9-fold differential transcription of ovine CCR5 and reduced proviral level of ovine progressive pneumonia virus. Anim. Genet. 2009, 40, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Larruskain, A.; Bernales, I.; Lujan, L.; de Andres, D.; Amorena, B.; Jugo, B.M. Expression analysis of 13 ovine immune response candidate genes in Visna/Maedi disease progression. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Alshanbari, F.A.; Mousel, M.R.; Reynolds, J.O.; Herrmann-Hoesing, L.M.; Highland, M.A.; Lewis, G.S.; White, S.N. Mutations in Ovis aries TMEM154 are associated with lower small ruminant lentivirus proviral concentration in one sheep flock. Anim. Genet. 2014, 45, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Cheevers, W.P.; McGuire, T.C. Equine infectious anemia virus: Immunopathogenesis and persistence. Rev. Infect. Dis. 1985, 7, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A.; Davis, E.; Carpenter, S. Endotoxin treatment of equine infectious anaemia virus-infected horse macrophage cultures decreases production of infectious virus. J. Gen. Virol. 1998, 79 Pt 4, 747–755. [Google Scholar] [CrossRef]
- Cook, S.J.; Cook, R.F.; Montelaro, R.C.; Issel, C.J. Differential responses of Equus caballus and Equus asinus to infection with two pathogenic strains of equine infectious anemia virus. Vet. Microbiol. 2001, 79, 93–109. [Google Scholar] [CrossRef]
- Patel, J.R.; Heldens, J.G.; Bakonyi, T.; Rusvai, M. Important mammalian veterinary viral immunodiseases and their control. Vaccine 2012, 30, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.K.; Montelaro, R.C. Lessons in AIDS vaccine development learned from studies of equine infectious, anemia virus infection and immunity. Viruses 2013, 5, 2963–2976. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chang, H.; Ge, M.; Lin, Y.; Wang, X.; Guo, W.; Wang, X. Development of antigen capture ELISA for the quantification of EIAV p26 protein. Appl. Microbiol. Biotechnol. 2014, 98, 9073–9081. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, K.; Cook, R.F.; Stefanetti, V.; Passamonti, F.; Autorino, G.L.; Scicluna, M.T.; Coletti, M.; Verini Supplizi, A.; Capomaccio, S. Deep sequencing and variant analysis of an Italian pathogenic field strain of equine infectious anaemia virus. Transbound. Emerg. Dis. 2017, 64, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Scicluna, M.T.; Autorino, G.L.; Nogarol, C.; Ricci, I.; Frontoso, R.; Rosone, F.; Nardini, R. Validation of an indirect ELISA employing a chimeric recombinant gag and env peptide for the serological diagnosis of equine infectious anemia. J. Virol. Methods 2018, 251, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Scicluna, M.T.; Issel, C.J.; Cook, F.R.; Manna, G.; Cersini, A.; Rosone, F.; Frontoso, R.; Caprioli, A.; Antognetti, V.; Autorino, G.L. Is a diagnostic system based exclusively on agar gel immunodiffusion adequate for controlling the spread of equine infectious anaemia? Vet. Microbiol. 2013, 165, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Issel, C.J.; Scicluna, M.T.; Cook, S.J.; Cook, R.F.; Caprioli, A.; Ricci, I.; Rosone, F.; Craigo, J.K.; Montelaro, R.C.; Autorino, G.L. Challenges and proposed solutions for more accurate serological diagnosis of equine infectious anaemia. Vet. Rec. 2013, 172, 210. [Google Scholar] [CrossRef] [PubMed]
- Capomaccio, S.; Willand, Z.A.; Cook, S.J.; Issel, C.J.; Santos, E.M.; Reis, J.K.; Cook, R.F. Detection, molecular characterization and phylogenetic analysis of full-length equine infectious anemia (EIAV) gag genes isolated from Shackleford Banks wild horses. Vet. Microbiol. 2012, 157, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Shen, R.X.; Zhu, Z.Y.; Deng, X.L.; Cao, X.Z.; Wang, X.F.; Ma, J.; Jiang, C.G.; Zhao, L.P.; Lv, X.L.; et al. An attenuated EIAV vaccine strain induces significantly different immune responses from its pathogenic parental strain although with similar in vivo replication pattern. Antivir. Res. 2011, 92, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Craigo, J.K.; Howe, L.; Steckbeck, J.D.; Cook, S.; Issel, C.; Montelaro, R.C. A live attenuated equine infectious anemia virus proviral vaccine with a modified S2 gene provides protection from detectable infection by intravenous virulent virus challenge of experimentally inoculated horses. J. Virol. 2003, 77, 7244–7253. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Ho, E.W.; Brown, M.L.; Yamamoto, J.K. Isolation of a T-lymphotropic virus from domestic cats with an immunodeficiency-like syndrome. Science 1987, 235, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Arjona, A.; Escolar, E.; Soto, I.; Barquero, N.; Martin, D.; Gomez-Lucia, E. Seroepidemiological survey of infection by feline leukemia virus and immunodeficiency virus in Madrid and correlation with some clinical aspects. J. Clin. Microbiol. 2000, 38, 3448–3449. [Google Scholar] [PubMed]
- Lee, J.; Malmberg, J.L.; Wood, B.A.; Hladky, S.; Troyer, R.; Roelke, M.; Cunningham, M.; McBride, R.; Vickers, W.; Boyce, W.; Boydston, E.; et al. Feline Immunodeficiency Virus Cross-Species Transmission: Implications for Emergence of New Lentiviral Infections. J. Virol. 2017, 91, e02134-16. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Tavares, L. Phylogenetic analysis of Portuguese Feline Immunodeficiency Virus sequences reveals high genetic diversity. Vet. Microbiol. 2006, 114, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, L.I.; Looney, D.J. Feline immunodeficiency virus: A concise review. Front. Biosci. 2004, 9, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, M.J.; Dean, G.A. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr. HIV Res. 2003, 1, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Hosie, M.J. The virus-receptor interaction in the replication of feline immunodeficiency virus (FIV). Curr. Opin. Virol. 2013, 3, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Arjona, A.; Barquero, N.; Domenech, A.; Tejerizo, G.; Collado, V.M.; Toural, C.; Martin, D.; Gomez-Lucia, E. Evaluation of a novel nested PCR for the routine diagnosis of feline leukemia virus (FeLV) and feline immunodeficiency virus (FIV). J. Feline Med. Surg. 2007, 9, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Westman, M.E.; Paul, A.; Malik, R.; McDonagh, P.; Ward, M.P.; Hall, E.; Norris, J.M. Seroprevalence of feline immunodeficiency virus and feline leukaemia virus in Australia: Risk factors for infection and geographical influences (2011–2013). JFMS Open Rep. 2016, 2, 2055116916646388. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, H.; Hohdatsu, T.; Okumura, M.; Sato, K.; Suzuki, Y.; Motokawa, K.; Gemma, T.; Watanabe, R.; Huang, C.; Arai, S.; et al. Dual-subtype vaccine (Fel-O-Vax FIV) protects cats against contact challenge with heterologous subtype B FIV infected cats. Vet. Microbiol. 2005, 108, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Brunt, J.; Guptill, L.; Kordick, D.L.; Kudrak, S.; Lappin, M.R.; American Association of Feline Practitioners; Academy of Feline Medicine Advisory Panel. American Association of Feline Practitioners 2006 Panel report on diagnosis, treatment, and prevention of Bartonella spp. infections. J. Feline Med. Surg. 2006, 8, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Bouillant, A.M.; Archambault, D. Bovine immunodeficiency virus: Short review. Ann. Rech. Vet. 1990, 21, 239–250. [Google Scholar] [PubMed]
- Kertayadnya, G.; Wilcox, G.E.; Soeharsono, S.; Hartaningsih, N.; Coelen, R.J.; Cook, R.D.; Collins, M.E.; Brownlie, J. Characteristics of a retrovirus associated with Jembrana disease in Bali cattle. J. Gen. Virol. 1993, 74 Pt 9, 1765–1778. [Google Scholar] [CrossRef]
- Yamada, E.; Yoshikawa, R.; Nakano, Y.; Misawa, N.; Kobayashi, T.; Ren, F.; Izumi, T.; Miyazawa, T.; Koyanagi, Y.; Sato, K. A naturally occurring bovine APOBEC3 confers resistance to bovine lentiviruses: Implication for the co-evolution of bovids and their lentiviruses. Sci. Rep. 2016, 6, 33988. [Google Scholar] [CrossRef] [PubMed]
- Desport, M.; Lewis, J. Jembrana disease virus: Host responses, viral dynamics and disease control. Curr. HIV Res. 2010, 8, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Patil, S.S.; Sood, R. Bovine immunodeficiency virus: A lentiviral infection. Indian J. Virol. 2013, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Passos-Castilho, A.M.; Marchand, C.; Archambault, D. B23/nucleophosmin interacts with bovine immunodeficiency virus Rev protein and facilitates viral replication. Virology 2018, 515, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wood, C.; Xue, W.; Krukenberg, S.M.; Chen, Q.; Minocha, H.C. Immune suppression in calves with bovine immunodeficiency virus. Clin. Diagn. Lab. Immunol. 1997, 4, 232–235. [Google Scholar] [PubMed]
- Corredor, A.G.; St-Louis, M.C.; Archambault, D. Molecular and biological aspects of the bovine immunodeficiency virus. Curr. HIV Res. 2010, 8, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, X.; Yao, X.; Kong, X.; Qiao, W.; Geng, Y. Bovine ISG15: An antiviral and inducible protein in BIV infected fetal bovine lung cells. Virol. J. 2010, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Ditcham, W.G.; Lewis, J.R.; Dobson, R.J.; Hartaningsih, N.; Wilcox, G.E.; Desport, M. Vaccination reduces the viral load and the risk of transmission of Jembrana disease virus in Bali cattle. Virology 2009, 386, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.; Derse, D. Physical and functional characterization of transcriptional control elements in the equine infectious anemia virus promoter. J. Virol. 1993, 67, 2064–2074. [Google Scholar] [PubMed]
- Shih, D.S.; Carruth, L.M.; Anderson, M.; Clements, J.E. Involvement of FOS and JUN in the activation of visna virus gene expression in macrophages through an AP-1 site in the viral LTR. Virology 1992, 190, 84–91. [Google Scholar] [CrossRef]
- Sparger, E.E.; Shacklett, B.L.; Renshaw-Gegg, L.; Barry, P.A.; Pedersen, N.C.; Elder, J.H.; Luciw, P.A. Regulation of gene expression directed by the long terminal repeat of the feline immunodeficiency virus. Virology 1992, 187, 165–177. [Google Scholar] [CrossRef]
- Crespo, H.; Bertolotti, L.; Juganaru, M.; Glaria, I.; de Andres, D.; Amorena, B.; Rosati, S.; Reina, R. Small ruminant macrophage polarization may play a pivotal role on lentiviral infection. Vet. Res. 2013, 44, 83. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Reina, R.; Glaria, I.; Ramirez, H.; de Andres, X.; Jauregui, P.; Lujan, L.; Martinez-Pomares, L.; Amorena, B.; de Andres, D.F. Identification of the ovine mannose receptor and its possible role in Visna/Maedi virus infection. Vet. Res. 2011, 42, 28. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Jauregui, P.; Glaria, I.; Sanjose, L.; Polledo, L.; Garcia-Marin, J.F.; Lujan, L.; de Andres, D.; Amorena, B.; Reina, R. Mannose receptor may be involved in small ruminant lentivirus pathogenesis. Vet. Res. 2012, 43, 43. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Ma, F.; Quinn, M.; Xiang, S.H. Genome-Wide Search for Host Association Factors during Ovine Progressive Pneumonia Virus Infection. PLoS ONE 2016, 11, e0150344. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Lowe, M.G.; Crowe, S.M.; O’Doherty, U.; Pope, M.; Gezelter, S.; Steinman, R.M. Susceptibility of dendritic cells to HIV-1 infection in vitro. J. Leukoc. Biol. 1994, 56, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Tiley, L.; McConnell, I.; Blacklaws, B. Infection of dendritic cells by the Maedi-Visna lentivirus. J. Virol. 2000, 74, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Kaba, J.; Winnicka, A.; Zaleska, M.; Nowicki, M.; Bagnicka, E. Influence of chronic caprine arthritis-encephalitis virus infection on the population of peripheral blood leukocytes. Pol. J. Vet. Sci. 2011, 14, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Haas, W.; Pereira, P.; Tonegawa, S. Gamma/delta cells. Annu. Rev. Immunol. 1993, 11, 637–685. [Google Scholar] [CrossRef] [PubMed]
- Jolly, P.E.; Gangopadhyay, A.; Chen, S.; Reddy, P.G.; Weiss, H.L.; Sapp, W.J. Changes in the leukocyte phenotype profile of goats infected with the caprine arthritis encephalitis virus. Vet. Immunol. Immunopathol. 1997, 56, 97–106. [Google Scholar] [CrossRef]
- Stonos, N.; Wootton, S.K.; Karrow, N. Immunogenetics of small ruminant lentiviral infections. Viruses 2014, 6, 3311–3333. [Google Scholar] [CrossRef] [PubMed]
- Mdurvwa, E.G.; Ogunbiyi, P.O.; Gakou, H.S.; Reddy, P.G. Pathogenic mechanisms of caprine arthritis-encephalitis virus. Vet. Res. Commun. 1994, 18, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Craigo, J.K.; Durkin, S.; Sturgeon, T.J.; Tagmyer, T.; Cook, S.J.; Issel, C.J.; Montelaro, R.C. Immune suppression of challenged vaccinates as a rigorous assessment of sterile protection by lentiviral vaccines. Vaccine 2007, 25, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Bolfa, P.F.; Leroux, C.; Pintea, A.; Andrei, S.; Catoi, C.; Taulescu, M.; Tabaran, F.; Spinu, M. Oxidant-antioxidant imbalance in horses infected with equine infectious anaemia virus. Vet. J. 2012, 192, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.D.; Na, L.; Zhu, C.H.; Shen, N.; Yang, F.; Fu, X.Q.; Wang, Y.H.; Fu, L.H.; Wang, J.Y.; Lin, Y.Z.; et al. Equine viperin restricts equine infectious anemia virus replication by inhibiting the production and/or release of viral Gag, Env, and receptor via distortion of the endoplasmic reticulum. J. Virol. 2014, 88, 12296–12310. [Google Scholar] [CrossRef] [PubMed]
- Willett, B.J.; Cannon, C.A.; Hosie, M.J. Upregulation of surface feline CXCR4 expression following ectopic expression of CCR5: Implications for studies of the cell tropism of feline immunodeficiency virus. J. Virol. 2002, 76, 9242–9252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Reckling, S.; Dean, G.A. Phenotypic and functional analysis of CD1a+ dendritic cells from cats chronically infected with feline immunodeficiency virus. Comp. Immunol. Microbiol. Infect. Dis. 2015, 42, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Gonda, M.A.; Braun, M.J.; Carter, S.G.; Kost, T.A.; Bess, J.W., Jr.; Arthur, L.O.; Van der Maaten, M.J. Characterization and molecular cloning of a bovine lentivirus related to human immunodeficiency virus. Nature 1987, 330, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.R.; Johnstone, P.; Brownlie, J. Investigation of the cellular tropism of bovine immunodeficiency-like virus. Res. Vet. Sci. 1998, 65, 33–40. [Google Scholar] [CrossRef]
- Wu, D.; Murakami, K.; Morooka, A.; Jin, H.; Inoshima, Y.; Sentsui, H. In vivo transcription of bovine leukemia virus and bovine immunodeficiency-like virus. Virus Res. 2003, 97, 81–87. [Google Scholar] [CrossRef]
- Wright, S.M.; Mleczko, A.; Coats, K.S. Bovine immunodeficiency virus expression in vitro is reduced in the presence of beta-chemokines, MIP-1alpha, MIP-1beta and RANTES. Vet. Res. Commun. 2002, 26, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Desport, M.; Ditcham, W.G.; Lewis, J.R.; McNab, T.J.; Stewart, M.E.; Hartaningsih, N.; Wilcox, G.E. Analysis of Jembrana disease virus replication dynamics in vivo reveals strain variation and atypical responses to infection. Virology 2009, 386, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shimizu, T. Toward a structural understanding of nucleic acid-sensing Toll-like receptors in the innate immune system. FEBS Lett. 2017, 591, 3167–3181. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Jungi, T.W.; Farhat, K.; Burgener, I.A.; Werling, D. Toll-like receptors in domestic animals. Cell Tissue Res. 2011, 343, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004, 25, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Mikula, I., Jr.; Pastorekova, S.; Mikula, I., Sr. Toll-like receptors in immune response to the viral infections. Acta Virol. 2010, 54, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Mikula, I.; Bhide, M.; Pastorekova, S.; Mikula, I. Characterization of ovine TLR7 and TLR8 protein coding regions, detection of mutations and Maedi Visna virus infection. Vet. Immunol. Immunopathol. 2010, 138, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Mikula, I., Jr.; Mikula, I., Sr. Characterization of ovine Toll-like receptor 9 protein coding region, comparative analysis, detection of mutations and maedi visna infection. Dev. Comp. Immunol. 2011, 35, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hickford, J.G. Polymorphism of Toll-like receptor 9 (TLR9) gene in sheep. Vet. Immunol. Immunopathol. 2008, 121, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Sarafidou, T.; Stamatis, C.; Kalozoumi, G.; Spyrou, V.; Fthenakis, G.C.; Billinis, C.; Mamuris, Z. Toll like receptor 9 (TLR9) polymorphism G520R in sheep is associated with seropositivity for Small Ruminant Lentivirus. PLoS ONE 2013, 8, e63901. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, S.S.; Lin, Y.Z.; Liu, H.F.; Liu, Q.; Wei, H.M.; Wang, X.F.; Wang, Y.H.; Du, C.; Kong, X.G.; et al. Infection of equine monocyte-derived macrophages with an attenuated equine infectious anemia virus (EIAV) strain induces a strong resistance to the infection by a virulent EIAV strain. Vet. Res. 2014, 45, 82. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, G.; Nordone, S.; Howard, K.E.; Dean, G.A. Toll-like receptor expression in feline lymphoid tissues. Vet. Immunol. Immunopathol. 2005, 106, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Cairns, K.; Kumar, S.; Leavell, S.; Assogba, B.D.; Burkhard, M.J. Effects of Alloantigen Exposure and Cell-Associated Mucosal FIV Challenge on Feline Toll-Like Receptor Gene Expression. In Proceedings of the ACVIM, San Antonio, TX, USA, 4–7 June 2008. [Google Scholar]
- Vahlenkamp, T.W.; Tompkins, M.B.; Tompkins, W.A. Feline immunodeficiency virus infection phenotypically and functionally activates immunosuppressive CD4+CD25+ T regulatory cells. J. Immunol. 2004, 172, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Martal, J.L.; Chene, N.M.; Huynh, L.P.; L’Haridon, R.M.; Reinaud, P.B.; Guillomot, M.W.; Charlier, M.A.; Charpigny, S.Y. IFN-tau: A novel subtype I IFN1. Structural characteristics, non-ubiquitous expression, structure-function relationships, a pregnancy hormonal embryonic signal and cross-species therapeutic potentialities. Biochimie 1998, 80, 755–777. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Takaoka, A.; Taniguchi, T. Regulation of the type I IFN induction: A current view. Int. Immunol. 2005, 17, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G.; Weber, F. The interferon response circuit: Induction and suppression by pathogenic viruses. Virology 2006, 344, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Benlahrech, A.; Gotch, F.; Kelleher, P.; Patterson, S. Loss of NK stimulatory capacity by plasmacytoid and monocyte-derived DC but not myeloid DC in HIV-1 infected patients. PLoS ONE 2011, 6, e17525. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med. 2011, 3, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Signal transduction and transcriptional regulation of interferon-alpha-stimulated genes. J. Interferon Res. 1991, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lucia, E.; Collado, V.M.; Miro, G.; Domenech, A. Effect of type-I interferon on retroviruses. Viruses 2009, 1, 545–573. [Google Scholar] [CrossRef] [PubMed]
- Zink, M.C.; Narayan, O. Lentivirus-induced interferon inhibits maturation and proliferation of monocytes and restricts the replication of caprine arthritis-encephalitis virus. J. Virol. 1989, 63, 2578–2584. [Google Scholar] [PubMed]
- Bertoni, G.B.B.A. Small Ruminant Lentiviruses and Cross Species Transmission. In Lentivirus and Macrophages: Molecular and Cellular Interactions; Caister Academic Press: Norfolk, UK, 2010. [Google Scholar]
- Detournay, O.; Morrison, D.A.; Wagner, B.; Zarnegar, B.; Wattrang, E. Genomic analysis and mRNA expression of equine type I interferon genes. J. Interferon Cytokine Res. 2013, 33, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, F.; Mauel, S.; Beier, I. Recombinant equine interferons: Expression cloning and biological activity. Vet. Immunol. Immunopathol. 2002, 84, 83–95. [Google Scholar] [CrossRef]
- Hu, Z.; Wu, X.; Ge, J.; Wang, X. Inhibition of virus replication and induction of human tetherin gene expression by equine IFN-alpha1. Vet. Immunol. Immunopathol. 2013, 156, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Robert-Tissot, C.; Ruegger, V.L.; Cattori, V.; Meli, M.L.; Riond, B.; Gomes-Keller, M.A.; Vogtlin, A.; Wittig, B.; Juhls, C.; Hofmann-Lehmann, R.; et al. The innate antiviral immune system of the cat: Molecular tools for the measurement of its state of activation. Vet. Immunol. Immunopathol. 2011, 143, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Domenech, A.; Miro, G.; Collado, V.M.; Ballesteros, N.; Sanjose, L.; Escolar, E.; Martin, S.; Gomez-Lucia, E. Use of recombinant interferon omega in feline retrovirosis: From theory to practice. Vet. Immunol. Immunopathol. 2011, 143, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S. The Use of Recombinant Feline Interferon Omega Therapy as an Immune-Modulator in Cats Naturally Infected with Feline Immunodeficiency Virus: New Perspectives. Vet. Sci. 2016, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S.; Duarte, A.; McGahie, D.; Sepulveda, N.; Niza, M.M.; Tavares, L. Evaluation of viremia, proviral load and cytokine profile in naturally feline immunodeficiency virus infected cats treated with two different protocols of recombinant feline interferon omega. Res. Vet. Sci. 2015, 99, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Collado, A.; Victorio, M. Efecto “In Vitro” de Interferón de Tipo I Sobre la Expresión de Retrovirus Felinos y Evaluación de su Aplicación Terapéutica en Gatos Con Infección Natural. Ph.D. Thesis, Universidad Complutense de Madrid, Madrid, Spain, May 2017. [Google Scholar]
- Chen, H.; Liu, X.; Li, Z.; Zhan, P.; De Clercq, E. TSG101: A novel anti-HIV-1 drug target. Curr. Med. Chem. 2010, 17, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.; Bieniasz, P. Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 2009, 29, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Munk, C. Cellular restriction factors of feline immunodeficiency virus. Viruses 2011, 3, 1986–2005. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kim, J.; Song, B.; Finzi, A.; Pacheco, B.; Sodroski, J. Virus-Specific Effects of TRIM5 Alpha(rh) RING Domain Functions on Restriction of Retroviruses. J. Virol. 2013, 87, 7234–7245. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, P.; Crespo, H.; Glaria, I.; Lujan, L.; Contreras, A.; Rosati, S.; de Andres, D.; Amorena, B.; Towers, G.J.; Reina, R. Ovine TRIM5alpha can restrict visna/maedi virus. J. Virol. 2012, 86, 9504–9509. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. Restriction factors: A defense against retroviral infection. Trends Microbiol. 2003, 11, 286–291. [Google Scholar] [CrossRef]
- Forshey, B.M.; Shi, J.; Aiken, C. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J. Virol. 2005, 79, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, S.; Grutter, C.; Strambio de Castillia, C.; Pertel, T.; Olivari, S.; Grutter, M.G.; Luban, J. An invariant surface patch on the TRIM5alpha PRYSPRY domain is required for retroviral restriction but dispensable for capsid binding. J. Virol. 2009, 83, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Chandrasekaran, V.; Carter, S.D.; Woodward, C.L.; Christensen, D.E.; Dryden, K.A.; Pornillos, O.; Yeager, M.; Ganser-Pornillos, B.K.; Jensen, G.J.; et al. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sastri, J.; Campbell, E.M. Recent insights into the mechanism and consequences of TRIM5alpha retroviral restriction. AIDS Res. Hum. Retrovir. 2011, 27, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Stremlau, M.; Yuan, W.; Song, B.; Perron, M.; Sodroski, J. Functional replacement of the RING, B-box 2, and coiled-coil domains of tripartite motif 5alpha (TRIM5alpha) by heterologous TRIM domains. J. Virol. 2006, 80, 6198–6206. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Si, Z.; Vandegraaff, N.; O’Huigin, C.; Song, B.; Yuan, W.; Xu, C.; Perron, M.; Li, X.; Marasco, W.A.; Engelman, A.; et al. Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc. Natl. Acad. Sci. USA 2006, 103, 7454–7459. [Google Scholar] [CrossRef] [PubMed]
- Ylinen, L.M.; Keckesova, Z.; Webb, B.L.; Gifford, R.J.; Smith, T.P.; Towers, G.J. Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J. Virol. 2006, 80, 7332–7338. [Google Scholar] [CrossRef] [PubMed]
- Poeschla, E.M. Primate and feline lentiviruses in current intrinsic immunity research: The cat is back. Vet. Immunol. Immunopathol. 2011, 143, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Wu, L.I.; Emerman, M.; Malik, H.S. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc. Natl. Acad. Sci. USA 2005, 102, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Sawyer, S.L. Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics 2009, 61, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Tareen, S.U.; Sawyer, S.L.; Malik, H.S.; Emerman, M. An expanded clade of rodent Trim5 genes. Virology 2009, 385, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Blikstad, C.; Shokeer, A.; Kurtovic, S.; Mannervik, B. Emergence of a novel highly specific and catalytically efficient enzyme from a naturally promiscuous glutathione transferase. Biochim. Biophys. Acta 2008, 1780, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Vogt, P. Historical introduction to the general properties of retroviruses. In Retroviruses; Coffin, J.M., Hughes, S.H., Varmus, H.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997; pp. 1–25. [Google Scholar]
- Song, B.; Gold, B.; O’Huigin, C.; Javanbakht, H.; Li, X.; Stremlau, M.; Winkler, C.; Dean, M.; Sodroski, J. The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J. Virol. 2005, 79, 6111–6121. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.F. A presentation of the differences between the sheep and goat genetic maps. Genet. Sel. Evol. 2005, 37 (Suppl. 1), S1–S10. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.C.; Li, W.H. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 2001, 68, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.; Huder, J.B.; Boni, J.; Schonmann, M.; Muhlherr, J.; Lutz, H.; Schupbach, J. Direct evidence for natural transmission of small-ruminant lentiviruses of subtype A4 from goats to sheep and vice versa. J. Virol. 2004, 78, 7518–7522. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Hue, S.; Schaller, T.; Pillay, D.; Towers, G.J. Hare TRIM5alpha restricts divergent retroviruses and exhibits significant sequence variation from closely related lagomorpha TRIM5 genes. J. Virol. 2010, 84, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Towers, G.J. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology 2007, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Cowan, S.; Goff, S.P.; Bieniasz, P.D.; Towers, G.J. Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J. 2003, 22, 385–394. [Google Scholar] [CrossRef] [PubMed]
- McEwan, W.A.; Schaller, T.; Ylinen, L.M.; Hosie, M.J.; Towers, G.J.; Willett, B.J. Truncation of TRIM5 in the Feliformia explains the absence of retroviral restriction in cells of the domestic cat. J. Virol. 2009, 83, 8270–8275. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, I.; McMonagle, E.L.; Petit, S.J.; Vijayakrishnan, S.; Logan, N.; Chan, C.N.; Towers, G.J.; Hosie, M.J.; Willett, B.J. Feline tetherin efficiently restricts release of feline immunodeficiency virus but not spreading of infection. J. Virol. 2011, 85, 5840–5852. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Tsuji-Kawahara, S.; Sakamoto, M.; Langlois, M.A.; Neuberger, M.S.; Rada, C.; Miyazawa, M. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J. Virol. 2008, 82, 10998–11008. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Holmes, R.K.; Malim, M.H. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006, 80, 8450–8458. [Google Scholar] [CrossRef] [PubMed]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lilley, C.E.; Yu, Q.; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Crawford, D.; Blouch, K.; Browne, E.P.; Kohli, R.M.; Ross, S.R. Different modes of retrovirus restriction by human APOBEC3A and APOBEC3G in vivo. PLoS Pathog. 2014, 10, e1004145. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, S.R.; Hache, G.; Stenglein, M.D.; Fahrenkrug, S.C.; Andresdottir, V.; Harris, R.S. Evolutionarily conserved and non-conserved retrovirus restriction activities of artiodactyl APOBEC3F proteins. Nucleic Acids Res. 2006, 34, 5683–5694. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, M.; Greeve, J. Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J. Gen. Virol. 2007, 88 Pt 12, 3270–3274. [Google Scholar] [CrossRef]
- Noguchi, C.; Hiraga, N.; Mori, N.; Tsuge, M.; Imamura, M.; Takahashi, S.; Fujimoto, Y.; Ochi, H.; Abe, H.; Maekawa, T.; et al. Dual effect of APOBEC3G on Hepatitis B virus. J. Gen. Virol. 2007, 88 Pt 2, 432–440. [Google Scholar] [CrossRef]
- Janahi, E.M.; McGarvey, M.J. The inhibition of hepatitis B virus by APOBEC cytidine deaminases. J Viral Hepat. 2013, 20, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Zhu, D.; Wang, C.; Su, C.; Ma, J.; Ma, J.; Wang, X. Core-binding factor subunit beta is not required for non-primate lentiviral Vif-mediated APOBEC3 degradation. J. Virol. 2014, 88, 12112–12122. [Google Scholar] [CrossRef] [PubMed]
- Konno, Y.; Nagaoka, S.; Kimura, I.; Yamamoto, K.; Kagawa, Y.; Kumata, R.; Aso, H.; Ueda, M.T.; Nakagawa, S.; Kobayashi, T.; et al. New World feline APOBEC3 potently controls inter-genus lentiviral transmission. Retrovirology 2018, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Zhang, Z.; Gertzen, C.G.W.; Haussinger, D.; Gohlke, H.; Munk, C. Identification of a Conserved Interface of Human Immunodeficiency Virus Type 1 and Feline Immunodeficiency Virus Vifs with Cullin 5. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Larue, R.S.; Lengyel, J.; Jonsson, S.R.; Andresdottir, V.; Harris, R.S. Lentiviral Vif degrades the APOBEC3Z3/APOBEC3H protein of its mammalian host and is capable of cross-species activity. J. Virol. 2010, 84, 8193–8201. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.R.; Stanley, D.J.; Hultquist, J.F.; Johnson, J.R.; Mietrach, N.; Binning, J.M.; Jonsson, S.R.; Barelier, S.; Newton, B.W.; Johnson, T.L.; et al. Lineage-Specific Viral Hijacking of Non-canonical E3 Ubiquitin Ligase Cofactors in the Evolution of Vif Anti-APOBEC3 Activity. Cell Rep. 2015, 11, 1236–1250. [Google Scholar] [CrossRef] [PubMed]
- De Pablo-Maiso, L.; Glaria, I.; Crespo, H.; Nistal-Villan, E.; Andresdottir, V.; de Andres, D.; Amorena, B.; Reina, R. Characterization of Ovine A3Z1 Restriction Properties against Small Ruminant Lentiviruses (SRLVs). Viruses 2017, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Bravo, I.G.; Marino, D.; Conrad, E.; Perkovic, M.; Battenberg, M.; Cichutek, K.; Munk, C. Restriction of equine infectious anemia virus by equine APOBEC3 cytidine deaminases. J. Virol. 2009, 83, 7547–7559. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Tallmadge, R.L.; Oaks, J.L.; Carpenter, S.; Cullen, B.R. Equine infectious anemia virus resists the antiretroviral activity of equine APOBEC3 proteins through a packaging-independent mechanism. J. Virol. 2008, 82, 11889–11901. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.D.; Zhang, X.; Na, L.; Wang, X.F.; Fu, L.H.; Zhu, C.H.; Wang, X.; Zhou, J.H. Double-stranded-RNA-specific adenosine deaminase 1 (ADAR1) is proposed to contribute to the adaptation of equine infectious anemia virus from horses to donkeys. Arch. Virol. 2016, 161, 2667–2672. [Google Scholar] [CrossRef] [PubMed]
- Radetskyy, R.; Daher, A.; Gatignol, A. ADAR1 and PKR, interferon stimulated genes with clashing effects on HIV-1 replication. Cytokine Growth Factor Rev. 2018, 40, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gu, Q.; Marino, D.; Lee, K.L.; Kong, I.K.; Haussinger, D.; Munk, C. Feline APOBEC3s, Barriers to Cross-Species Transmission of FIV? Viruses 2018, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Marino, D.; Hofmann, H.; Yuhki, N.; Lochelt, M.; Munk, C. Vif of feline immunodeficiency virus from domestic cats protects against APOBEC3 restriction factors from many felids. J. Virol. 2010, 84, 7312–7324. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, H.; Zhou, X.; Li, Z.; Zheng, B.; Zhang, W. Jembrana disease virus Vif antagonizes the inhibition of bovine APOBEC3 proteins through ubiquitin-mediate protein degradation. Virology 2018, 519, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, H.; Li, Z.; Liu, X.; Liu, G.; Harris, R.S.; Yu, X.F. Cellular requirements for bovine immunodeficiency virus Vif-mediated inactivation of bovine APOBEC3 proteins. J. Virol. 2014, 88, 12528–12540. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Hu, Z.; Gu, Q.; Wu, X.; Zheng, Y.H.; Wei, P.; Wang, X. Equine tetherin blocks retrovirus release and its activity is antagonized by equine infectious anemia virus envelope protein. J. Virol. 2014, 88, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, J.; Qu, M.; Li, X.; Zhang, J.; Zhang, H.; Wu, J.; Yu, B.; Wu, H.; Kong, W.; et al. Viral Restriction Activity of Feline BST2 Is Independent of Its N-Glycosylation and Induction of NF-kappaB Activation. PLoS ONE 2015, 10, e0138190. [Google Scholar]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.M.N.; Aschman, N.; Renesto, P.; Usami, Y.; Gottlinger, H.G. Structural basis of tetherin function. Curr. HIV Res. 2012, 10, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.; Guatelli, J. Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J. Virol. 2013, 87, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, F.; Black, S.G.; Murphy, L.; Griffiths, D.J.; Neil, S.J.; Spencer, T.E.; Palmarini, M. Interplay between ovine bone marrow stromal cell antigen 2/tetherin and endogenous retroviruses. J. Virol. 2010, 84, 4415–4425. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Varela, M.; Desloire, S.; Ftaich, N.; Murgia, C.; Golder, M.; Neil, S.; Spencer, T.E.; Wootton, S.K.; Lavillette, D.; et al. The sheep tetherin paralog oBST2B blocks envelope glycoprotein incorporation into nascent retroviral virions. J. Virol. 2015, 89, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Sanjose, L.; Crespo, H.; Blatti-Cardinaux, L.; Glaria, I.; Martinez-Carrasco, C.; Berriatua, E.; Amorena, B.; De Andres, D.; Bertoni, G.; Reina, R. Post-entry blockade of small ruminant lentiviruses by wild ruminants. Vet. Res. 2016, 47, 1. [Google Scholar] [CrossRef] [PubMed]
- Crespo, H.; Bertolotti, L.; Proffiti, M.; Cascio, P.; Cerruti, F.; Acutis, P.L.; de Andres, D.; Reina, R.; Rosati, S. Low proviral small ruminant lentivirus load as biomarker of natural restriction in goats. Vet. Microbiol. 2016, 192, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Ma, J.; Wang, X.; Guo, M.; Li, Y.; Wang, X. A pilot study on interaction between donkey tetherin and EIAV stains with different virulent and replication characteristics. Microb. Pathog. 2017, 106, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, E.A.; Brennan, G.; Ferguson, B.; Wiseman, R.W.; O’Connor, D.; Hu, S.L. Variable prevalence and functional diversity of the antiretroviral restriction factor TRIMCyp in Macaca fascicularis. J. Virol. 2011, 85, 9956–9963. [Google Scholar] [CrossRef] [PubMed]
- Celestino, M.; Calistri, A.; Del Vecchio, C.; Salata, C.; Chiuppesi, F.; Pistello, M.; Borsetti, A.; Palu, G.; Parolin, C. Feline tetherin is characterized by a short N-terminal region and is counteracted by the feline immunodeficiency virus envelope glycoprotein. J. Virol. 2012, 86, 6688–6700. [Google Scholar] [CrossRef] [PubMed]
- Fukuma, A.; Abe, M.; Morikawa, Y.; Miyazawa, T.; Yasuda, J. Cloning and characterization of the antiviral activity of feline Tetherin/BST-2. PLoS ONE 2011, 6, e18247. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, I.; Hosie, M.J.; Willett, B.J. The role of BST2/tetherin in feline retrovirus infection. Vet. Immunol. Immunopathol. 2011, 143, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Guevara, R.B.; Marcano, A.C.; Saenz, D.T.; Fadel, H.J.; Rogstad, D.K.; Poeschla, E.M. Feline immunodeficiency virus envelope glycoproteins antagonize tetherin through a distinctive mechanism that requires virion incorporation. J. Virol. 2014, 88, 3255–3272. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Nakagawa, S.; Nakaya, Y.; Tanaka, A.; Miyazawa, T.; Yasuda, J. Identification and functional analysis of three isoforms of bovine BST-2. PLoS ONE 2012, 7, e41483. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Zhang, Y.; Song, J.; Zhang, H.; Zhang, S.; Li, Y.; Tan, J.; Qiao, W. The effect of bovine BST2A1 on the release and cell-to-cell transmission of retroviruses. Virol. J. 2017, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Qiao, F.; Bowie, J.U. The many faces of SAM. Sci. STKE 2005, 2005, re7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Greene, W.C. A new activity for SAMHD1 in HIV restriction. Nat. Med. 2014, 20, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Choi, J.; Oh, C.; Kim, S.; Seo, M.; Kim, S.Y.; Seo, D.; Kim, J.; White, T.E.; Brandariz-Nunez, A.; et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014, 20, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, J.; Hwang, S.Y.; Choi, J.; Oh, C.; Ahn, K. Reply to SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1074–1075. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; St Gelais, C.; de Silva, S.; Yount, J.S.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Kodigepalli, K.M.; Barrett, B.S.; Kim, S.H.; Antonucci, J.M.; Ladner, K.J.; Buzovetsky, O.; et al. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc. Natl. Acad. Sci. USA 2018, 115, E3798–E3807. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Kim, S.H.; St Gelais, C.; Bonifati, S.; Li, T.W.; Buzovetsky, O.; Knecht, K.M.; Duchon, A.A.; Xiong, Y.; Musier-Forsyth, K.; et al. SAMHD1 Impairs HIV-1 Gene Expression and Negatively Modulates Reactivation of Viral Latency in CD4(+) T Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.A.; Nguyen, V.T.; Fraldi, A.; Labas, V.; Edwards, M.; Bonnet, F.; Lania, L.; Bensaude, O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell. Biol. 2003, 23, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Price, J.P.; Byers, S.A.; Cheng, D.; Peng, J.; Price, D.H. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 2005, 280, 28819–28826. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Varrone, F.; Napolitano, G.; Michels, A.A.; Majello, B.; Bensaude, O.; Lania, L. Inhibition of Tat activity by the HEXIM1 protein. Retrovirology 2005, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.Y.; Ma, Y.G.; Gai, Y.M.; Liang, Z.B.; Ma, J.; Su, Y.; Zhang, Q.C.; Chen, Q.M.; Tan, J. Bovine HEXIM1 inhibits bovine immunodeficiency virus replication through regulating BTat-mediated transactivation. Vet. Res. 2013, 44, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.Z.; Sun, L.K.; Zhu, D.T.; Hu, Z.; Wang, X.F.; Du, C.; Wang, Y.H.; Wang, X.J.; Zhou, J.H. Equine schlafen 11 restricts the production of equine infectious anemia virus via a codon usage-dependent mechanism. Virology 2016, 495, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Pablo-Maiso, L.; Doménech, A.; Echeverría, I.; Gómez-Arrebola, C.; De Andrés, D.; Rosati, S.; Gómez-Lucia, E.; Reina, R. Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses. Viruses 2018, 10, 435. https://doi.org/10.3390/v10080435
De Pablo-Maiso L, Doménech A, Echeverría I, Gómez-Arrebola C, De Andrés D, Rosati S, Gómez-Lucia E, Reina R. Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses. Viruses. 2018; 10(8):435. https://doi.org/10.3390/v10080435
Chicago/Turabian StyleDe Pablo-Maiso, Lorena, Ana Doménech, Irache Echeverría, Carmen Gómez-Arrebola, Damián De Andrés, Sergio Rosati, Esperanza Gómez-Lucia, and Ramsés Reina. 2018. "Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses" Viruses 10, no. 8: 435. https://doi.org/10.3390/v10080435
APA StyleDe Pablo-Maiso, L., Doménech, A., Echeverría, I., Gómez-Arrebola, C., De Andrés, D., Rosati, S., Gómez-Lucia, E., & Reina, R. (2018). Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses. Viruses, 10(8), 435. https://doi.org/10.3390/v10080435