Functional Genomics and Immunologic Tools: The Impact of Viral and Host Genetic Variations on the Outcome of Zika Virus Infection

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Sequence Alignment and Phylogenetic Analysis
2.3. Cloning
2.4. Transcription and Transfection
2.5. Growth Kinetics and Cytopathogenicity
2.6. Real-Time RT-PCR
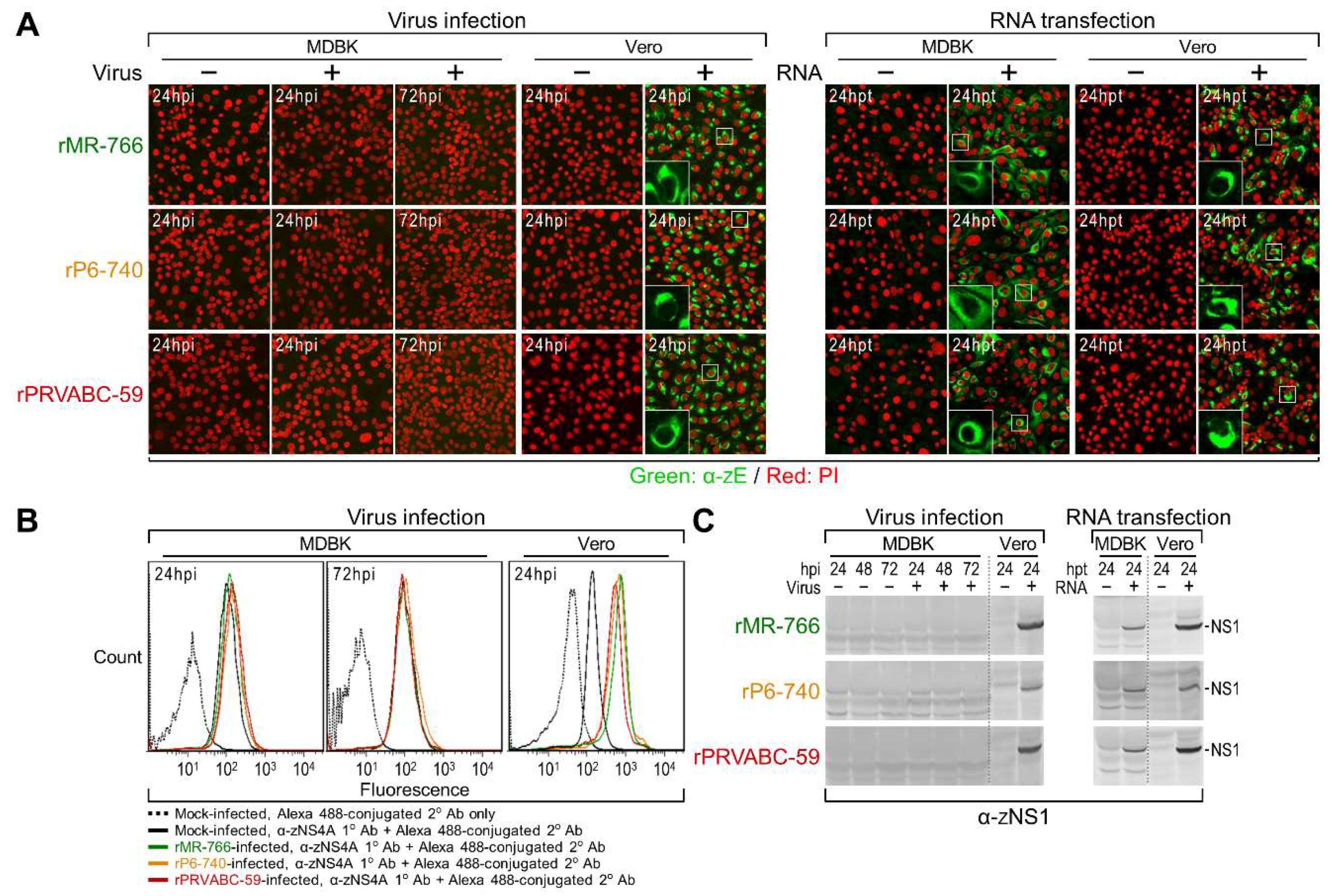
2.7. Immunoblotting, Confocal Microscopy, and Flow Cytometry
2.8. Mouse Studies
2.9. Ethics Statement
3. Results
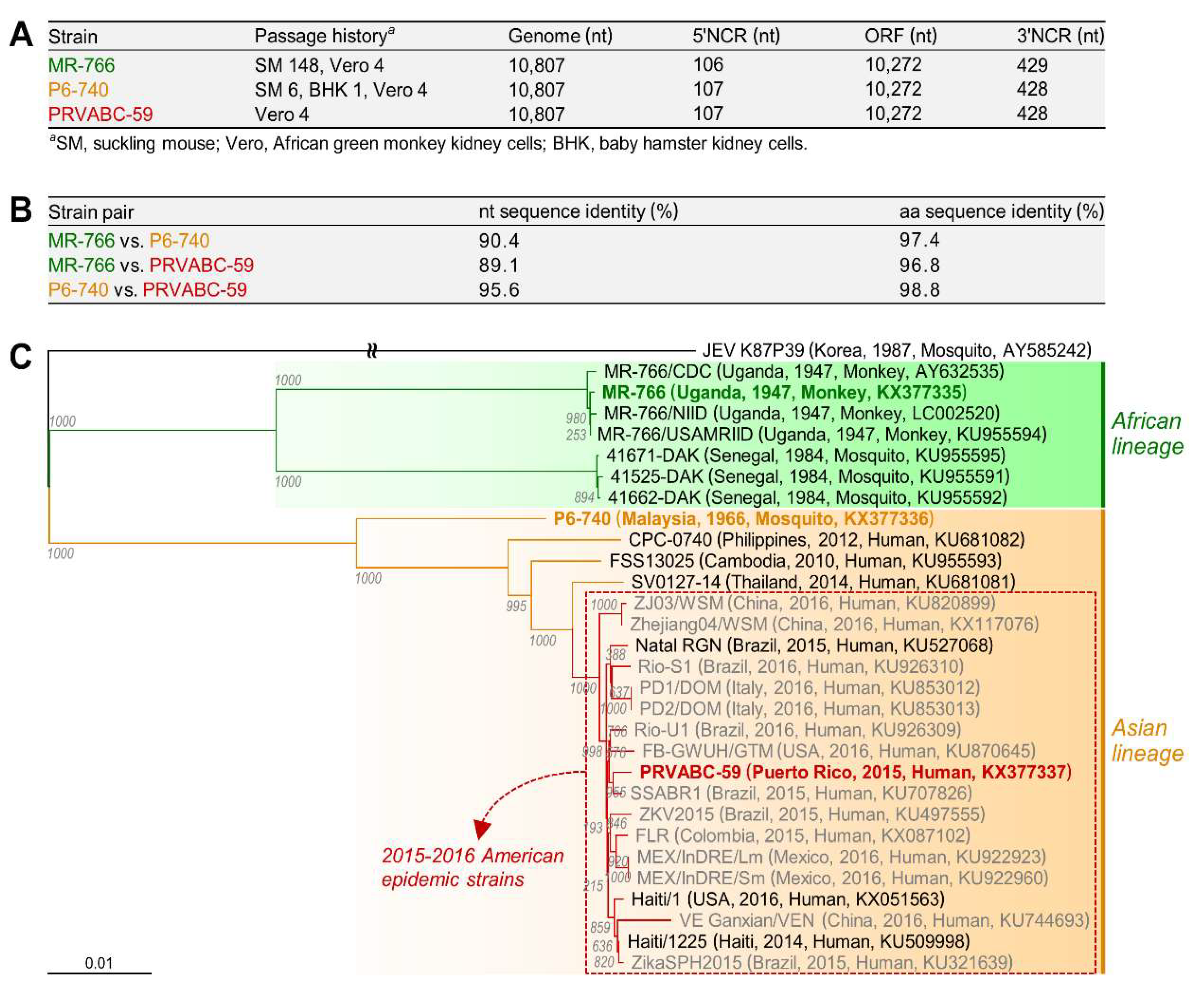
3.1. Characterization of Three Spatiotemporally Distinct and Genetically Divergent ZIKV Strains
3.2. Development of Genetically Stable Full-Length Infectious cDNA Clones for the Three ZIKV Strains
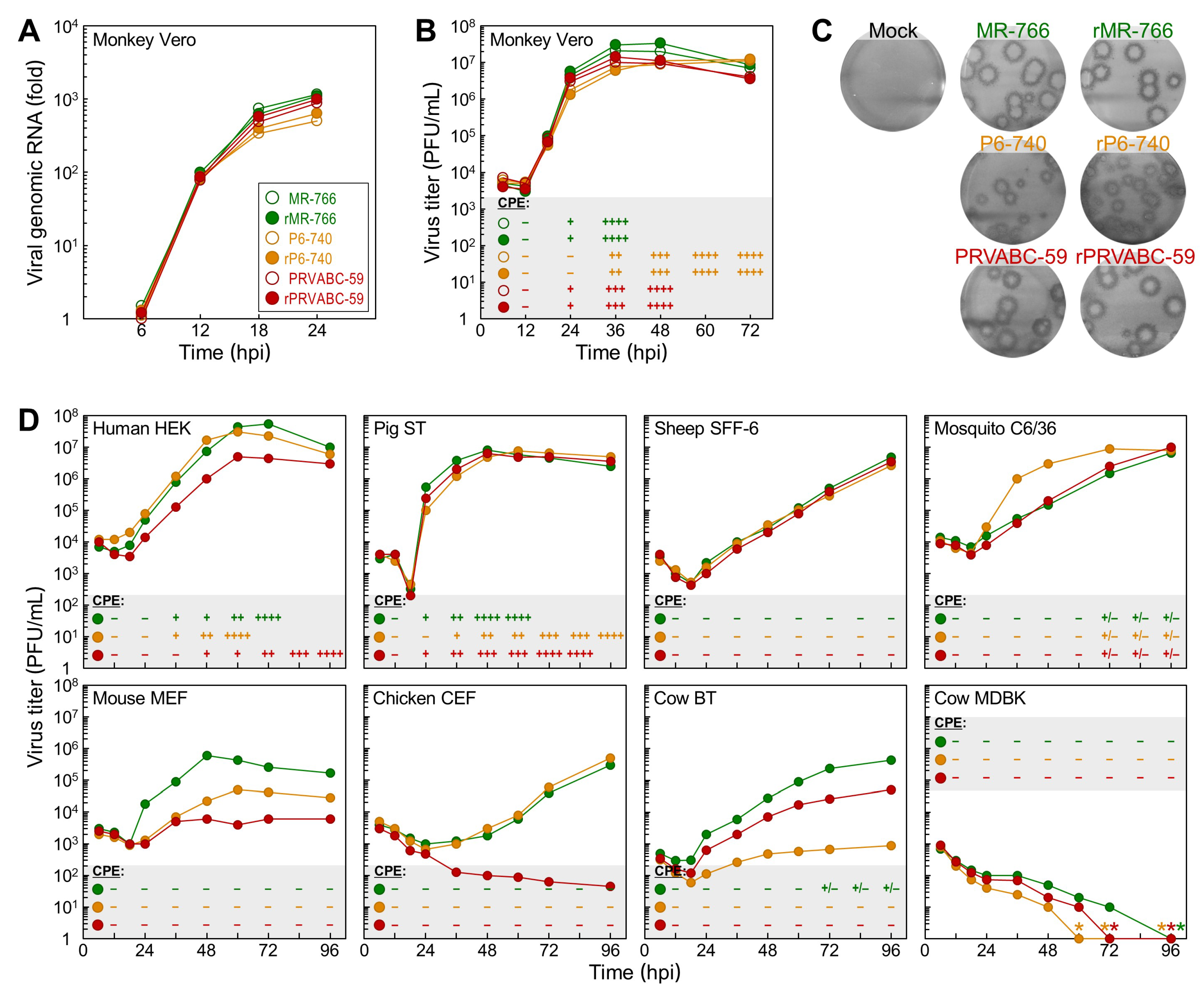
3.3. Differential Replication Kinetics and Cytopathogenicity among Three Molecularly Cloned ZIKVs in Human, Mosquito, and Animal Cell Lines
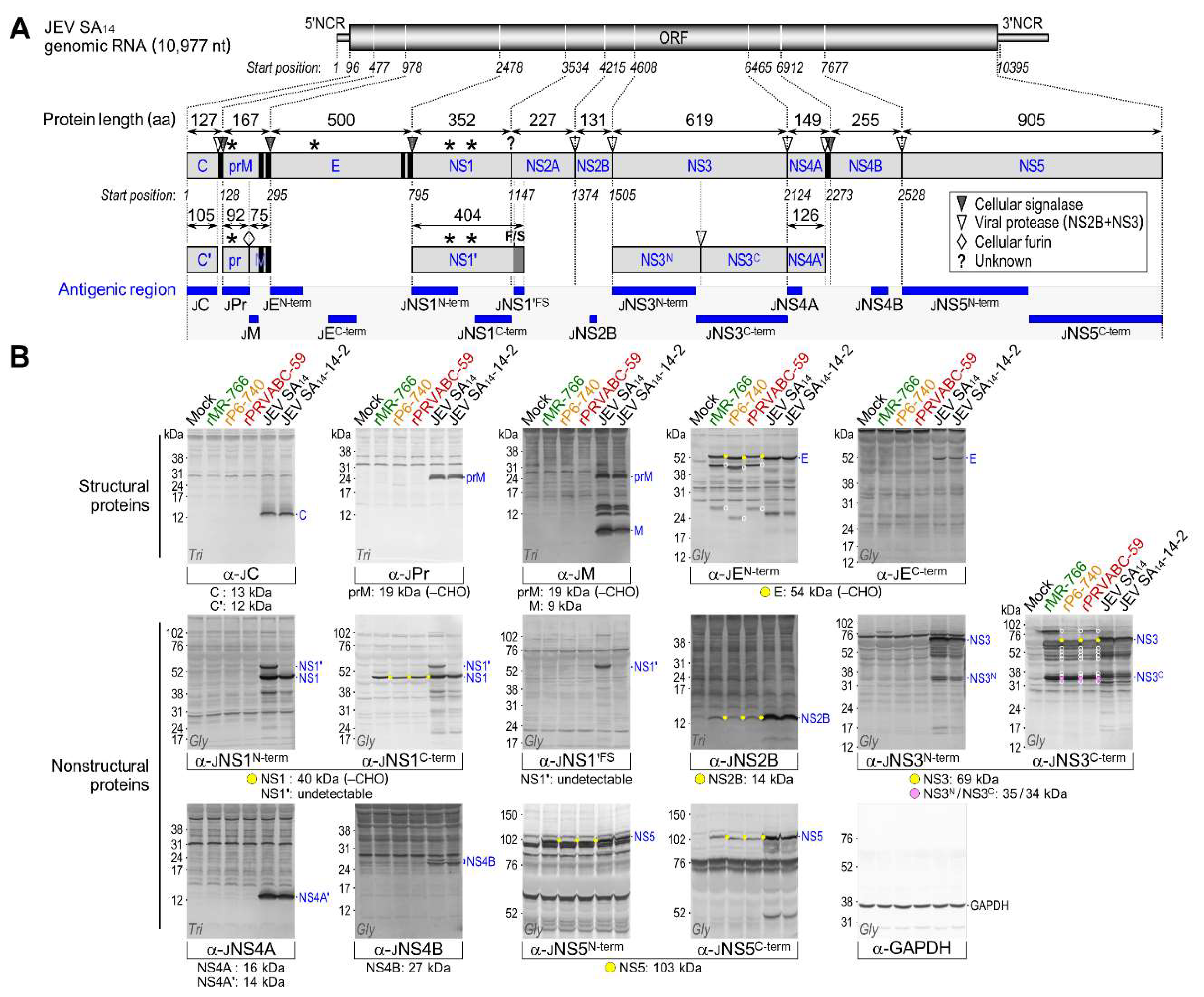
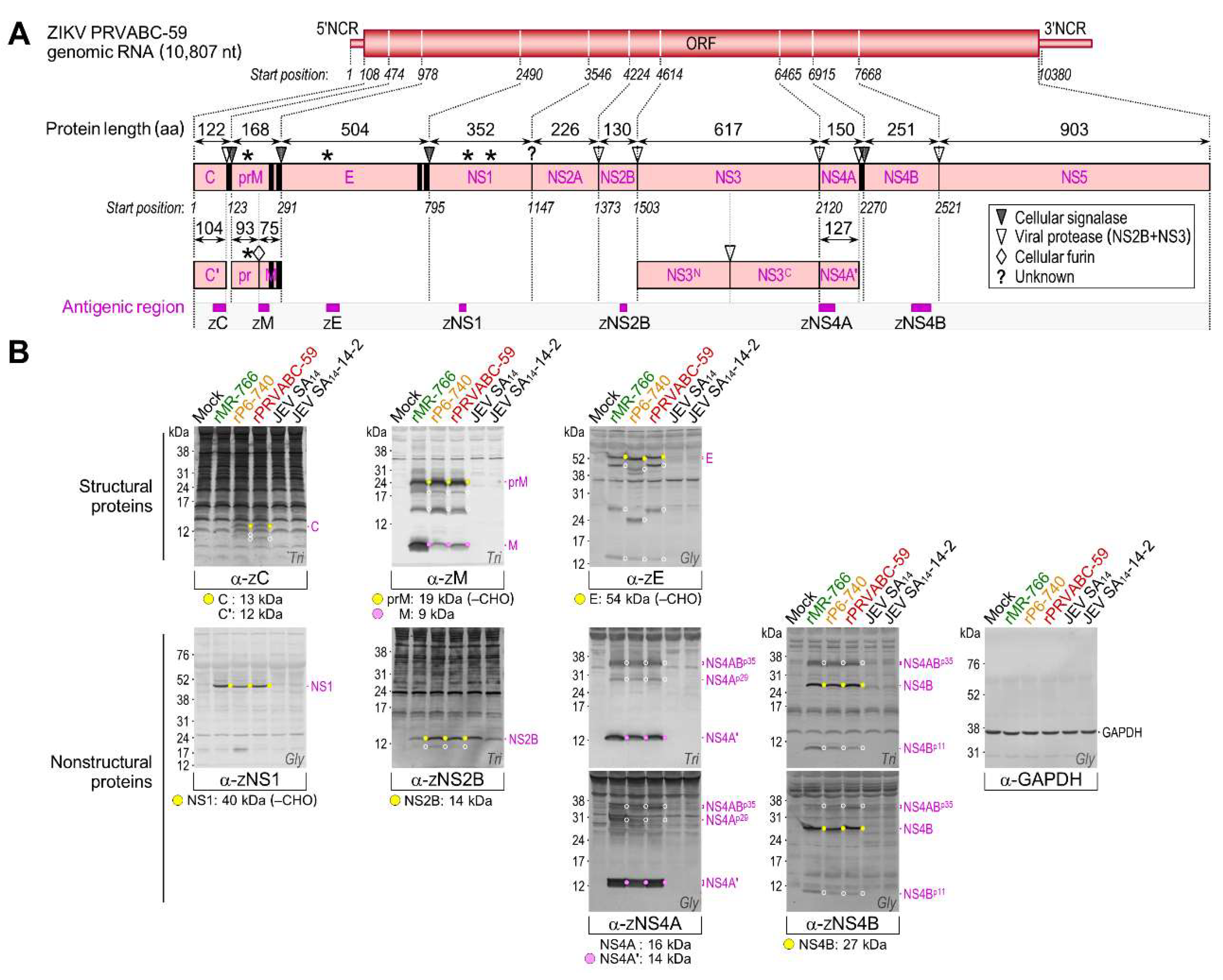
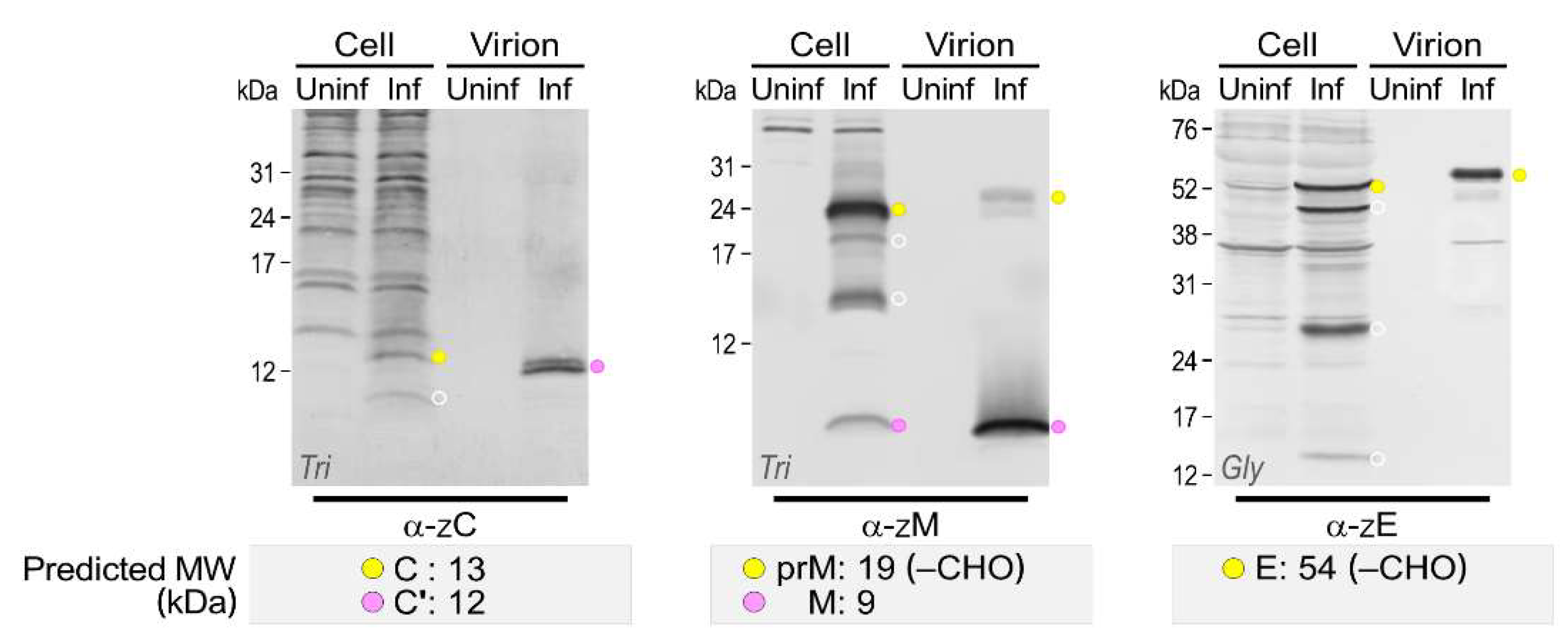
3.4. Genome-Wide Landscape of the Viral Gene Products and Their Related Species Produced by the Molecularly Cloned ZIKVs
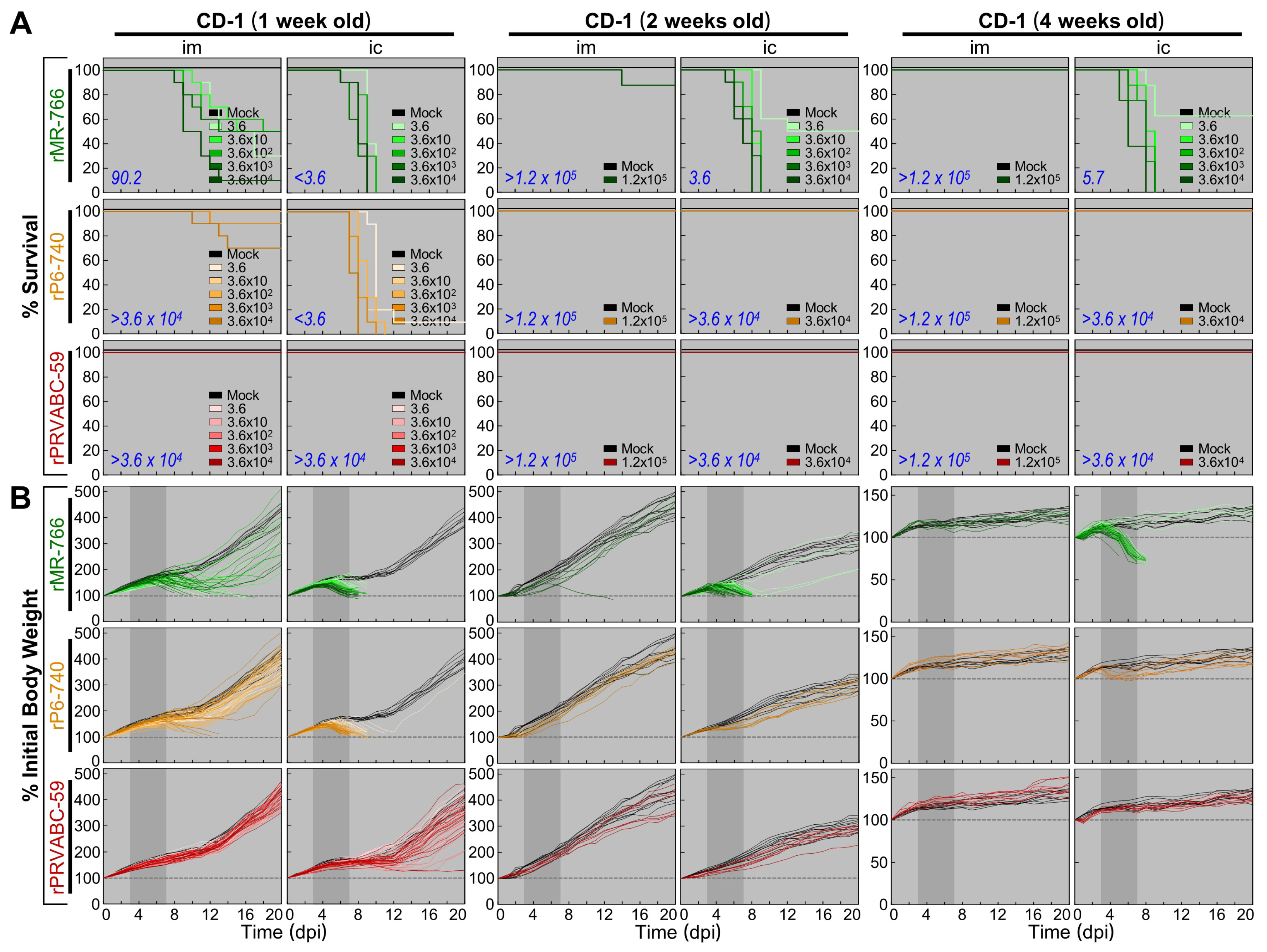
3.5. Wide Range of Differences in Age-Dependent Neuropathogenicity among Three Molecularly Cloned ZIKVs in Outbred CD-1 Mice
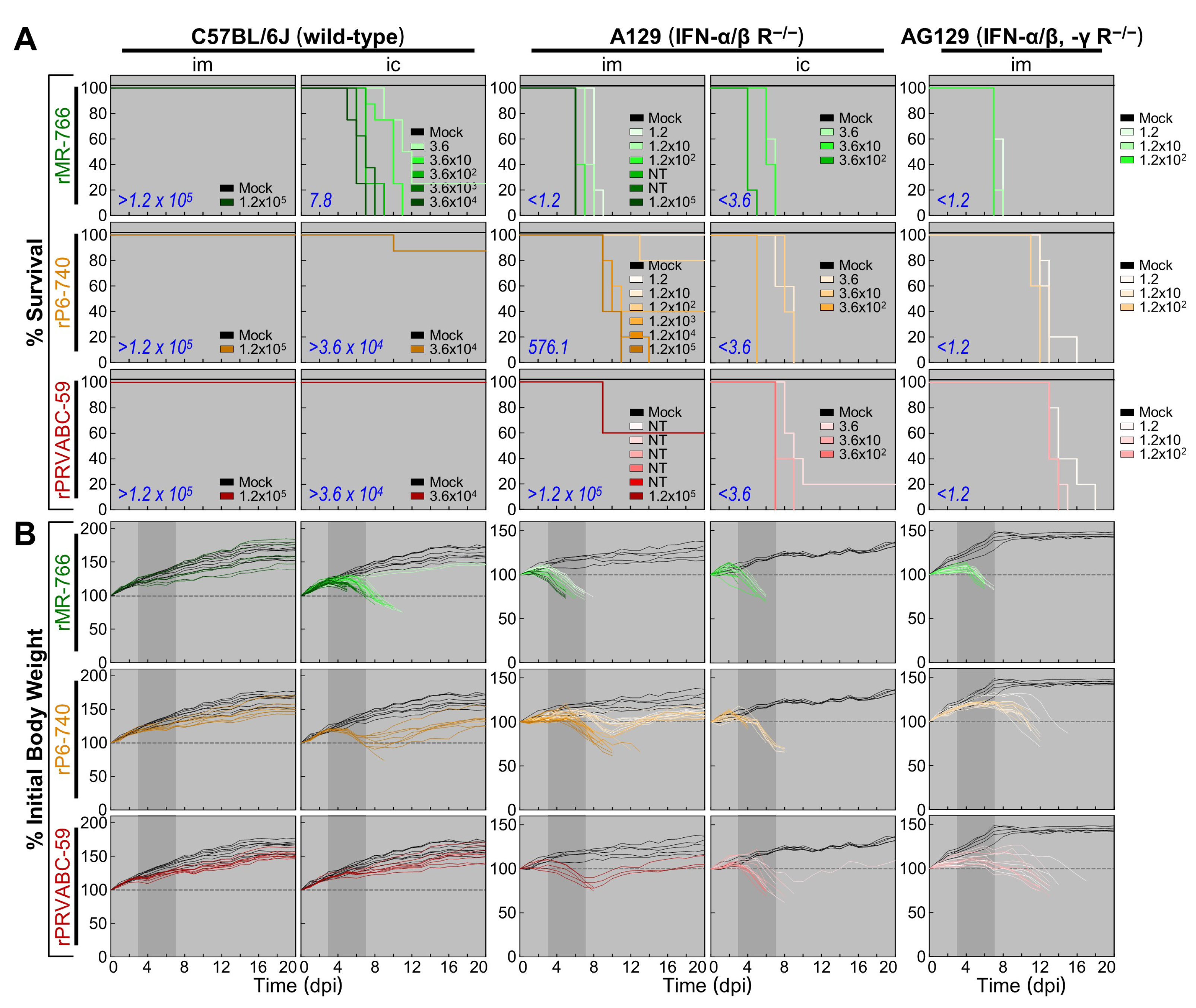
3.6. High Degree of Variation in Interferon (IFN) Sensitivity among Three Molecularly Cloned ZIKVs in Mice Lacking Type I (IFNAR−/−) or Both Type I and II IFN (IFNAR−/−/IFNGR−/−) Receptors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Lee, Y.M. Zika virus: An emerging flavivirus. J. Microbiol. 2017, 55, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Gubler, D.J. Zika virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Musso, D. Emerging arboviruses in the Pacific. Lancet 2014, 384, 1571–1572. [Google Scholar] [CrossRef]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika virus outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; Melo, V.C.; Mosimann, A.L.; Santos, G.I.; Santos, C.N.; Luz, K. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Lessler, J.; Chaisson, L.H.; Kucirka, L.M.; Bi, Q.; Grantz, K.; Salje, H.; Carcelen, A.C.; Ott, C.T.; Sheffield, J.S.; Ferguson, N.M.; et al. Assessing the global threat from Zika virus. Science 2016, 353, aaf8160. [Google Scholar] [CrossRef] [PubMed]
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.; de Sequeira, P.C.; de Mendonca, M.C.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Roche, C.; Robin, E.; Nhan, T.; Teissier, A.; Cao-Lormeau, V.M. Potential sexual transmission of Zika virus. Emerg. Infect. Dis. 2015, 21, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the Western Hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef] [PubMed]
- Panchaud, A.; Stojanov, M.; Ammerdorffer, A.; Vouga, M.; Baud, D. Emerging role of Zika virus in adverse fetal and neonatal outcomes. Clin. Microbiol. Rev. 2016, 29, 659–694. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Lim, E.X.; Zhang, S.; Fibriansah, G.; Ng, T.S.; Ooi, J.S.; Shi, J.; Lok, S.M. Structure of the thermally stable Zika virus. Nature 2016, 533, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 A resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Murray, C.L.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer Health: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus entry receptors: An update. Viruses 2014, 6, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Kielian, M. Flaviviruses: Braking the entering. Curr. Opin. Virol. 2013, 3, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Kuhn, R.J. Zika virus structure, maturation, and receptors. J. Infect. Dis. 2017, 216 (Suppl. 10), S935–S944. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Rossmann, M.G. Molecular mechanisms involved in the early steps of flavivirus cell entry. Microbes Infect. 2011, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, S.M.; Harris, E. Control of dengue virus translation and replication. Curr. Top. Microbiol. Immunol. 2010, 338, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, L.G.; Filomatori, C.V.; Gamarnik, A.V. Functional RNA elements in the dengue virus genome. Viruses 2011, 3, 1739–1756. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A. Replication cycle and molecular biology of the West Nile virus. Viruses 2014, 6, 13–53. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, J.M.; Song, B.H.; Yun, S.I.; Yun, G.N.; Byun, S.J.; Lee, Y.M. Profiling of viral proteins expressed from the genomic RNA of Japanese encephalitis virus using a panel of 15 region-specific polyclonal rabbit antisera: Implications for viral gene expression. PLoS ONE 2015, 10, e0124318. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Li, X.F.; Zhao, H.; Li, S.H.; Deng, Y.Q.; Cao, R.Y.; Song, K.Y.; Wang, H.J.; Hua, R.H.; Yu, Y.X.; et al. A single nucleotide mutation in NS2A of Japanese encephalitis-live vaccine virus (SA14-14-2) ablates NS1’ formation and contributes to attenuation. J. Gen. Virol. 2012, 93, 1959–1964. [Google Scholar] [CrossRef] [PubMed]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. NS1’ of flaviviruses in the Japanese encephalitis virus serogroup is a product of ribosomal frameshifting and plays a role in viral neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Atkins, J.F. A conserved predicted pseudoknot in the NS2A-encoding sequence of West Nile and Japanese encephalitis flaviviruses suggests NS1’ may derive from ribosomal frameshifting. Virol. J. 2009, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antiviral Res. 2010, 87, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. Degrees of maturity: The complex structure and biology of flaviviruses. Curr. Opin. Virol. 2012, 2, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Methot, P.O.; Alizon, S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence 2014, 5, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Haddow, A.D.; Schuh, A.J.; Yasuda, C.Y.; Kasper, M.R.; Heang, V.; Huy, R.; Guzman, H.; Tesh, R.B.; Weaver, S.C. Genetic characterization of Zika virus strains: Geographic expansion of the Asian lineage. PLoS Negl. Trop. Dis. 2012, 6, e1477. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Azevedo Rdo, S.; Kraemer, M.U.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Valderramos, S.G.; Wu, A.; Ouyang, S.; Li, C.; Brasil, P.; Bonaldo, M.; Coates, T.; Nielsen-Saines, K.; Jiang, T.; et al. From mosquitos to humans: Genetic evolution of Zika virus. Cell Host Microbe 2016, 19, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.E.; Diamond, M.S. Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol. 2017, 91, e00009-17. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a novel murine model to study Zika virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of lethal Zika virus infection in AG129 mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Sene, A.; Richner, J.M.; Smith, A.M.; Santeford, A.; Ban, N.; Weger-Lucarelli, J.; Manzella, F.; Ruckert, C.; Govero, J.; et al. Zika virus infection in mice causes panuveitis with shedding of virus in tears. Cell Rep. 2016, 16, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G.; Siddharthan, V.; Evans, J.; Taylor, R.; Tolbert, K.; Apuli, C.; Stewart, J.; Collins, P.; Gebre, M.; Neilson, S.; et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antiviral Res. 2017, 137, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.; Neyts, J. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef] [PubMed]
- Weger-Lucarelli, J.; Duggal, N.K.; Bullard-Feibelman, K.; Veselinovic, M.; Romo, H.; Nguyen, C.; Ruckert, C.; Brault, A.C.; Bowen, R.A.; Stenglein, M.; et al. Development and characterization of recombinant virus generated from a New World Zika virus infectious clone. J. Virol. 2017, 91, e01765-16. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Kim, S.Y.; Choi, W.Y.; Nam, J.H.; Ju, Y.R.; Park, K.Y.; Cho, H.W.; Lee, Y.M. Molecular characterization of the full-length genome of the Japanese encephalitis viral strain K87P39. Virus Res. 2003, 96, 129–140. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Yun, S.I.; Song, B.H.; Kim, J.K.; Lee, Y.M. Bacterial artificial chromosomes: A functional genomics tool for the study of positive-strand RNA viruses. J. Vis. Exp. 2015, 106, e53164. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Kim, S.Y.; Rice, C.M.; Lee, Y.M. Development and application of a reverse genetics system for Japanese encephalitis virus. J. Virol. 2003, 77, 6450–6465. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Song, B.H.; Frank, J.C.; Julander, J.G.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.M. Complete genome sequences of three historically important, spatiotemporally distinct, and genetically divergent strains of Zika virus: MR-766, P6-740, and PRVABC-59. Genome Announc. 2016, 4, e00800-16. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Yun, S.I.; Song, B.H.; Hahn, Y.S.; Lee, C.H.; Oh, H.W.; Lee, Y.M. A single N-linked glycosylation site in the Japanese encephalitis virus prM protein is critical for cell type-specific prM protein biogenesis, virus particle release, and pathogenicity in mice. J. Virol. 2008, 82, 7846–7862. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Choi, Y.J.; Song, B.H.; Lee, Y.M. 3′ cis-acting elements that contribute to the competence and efficiency of Japanese encephalitis virus genome replication: Functional importance of sequence duplications, deletions, and substitutions. J. Virol. 2009, 83, 7909–7930. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Song, B.H.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.M. Comparison of the live-attenuated Japanese encephalitis vaccine SA14-14-2 strain with its pre-attenuated virulent parent SA14 strain: Similarities and differences in vitro and in vivo. J. Gen. Virol. 2016, 97, 2575–2591. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.I.; Song, B.H.; Kim, J.K.; Yun, G.N.; Lee, E.Y.; Li, L.; Kuhn, R.J.; Rossmann, M.G.; Morrey, J.D.; Lee, Y.M. A molecularly cloned, live-attenuated Japanese encephalitis vaccine SA14-14-2 virus: A conserved single amino acid in the ij hairpin of the viral E glycoprotein determines neurovirulence in mice. PLoS Pathog. 2014, 10, e1004290. [Google Scholar] [CrossRef] [PubMed]
- Marchette, N.J.; Garcia, R.; Rudnick, A. Isolation of Zika virus from Aedes aegypti mosquitoes in Malaysia. Am. J. Trop. Med. Hyg. 1969, 18, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Lambert, A.J.; Holodniy, M.; Saavedra, S.; Signor Ldel, C. Phylogeny of Zika virus in Western Hemisphere, 2015. Emerg. Infect. Dis. 2016, 22, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yi, G.; Du, F.; Chuang, Y.C.; Vaughan, R.C.; Sankaran, B.; Kao, C.C.; Li, P. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 2017, 8, 14762. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tan, X.F.; Thurmond, S.; Zhang, Z.M.; Lin, A.; Hai, R.; Song, J. The structure of Zika virus NS5 reveals a conserved domain conformation. Nat. Commun. 2017, 8, 14763. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl mediates Zika virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.M.; Miller, A.S.; Klose, T.; Sirohi, D.; Buda, G.; Jiang, W.; Kuhn, R.J.; Rossmann, M.G. Structure of the immature Zika virus at 9 A resolution. Nat. Struct. Mol. Biol. 2017, 24, 184–186. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.C.; Akey, D.L.; Konwerski, J.R.; Tarrasch, J.T.; Skiniotis, G.; Kuhn, R.J.; Smith, J.L. Extended surface for membrane association in Zika virus NS1 structure. Nat. Struct. Mol. Biol. 2016, 23, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, H.; Qi, J.; Liu, Y.; Wang, H.; Su, C.; Shi, Y.; Gao, G.F. Contribution of intertwined loop to membrane association revealed by Zika virus full-length NS1 structure. EMBO J. 2016, 35, 2170–2178. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Setoh, Y.X.; Hall, R.A.; Khromykh, A.A. Post-Translational regulation and modifications of flavivirus structural proteins. J. Gen. Virol. 2015, 96, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Stobart, C.C.; Moore, M.L. RNA virus reverse genetics and vaccine design. Viruses 2014, 6, 2531–2550. [Google Scholar] [CrossRef] [PubMed]
- Aubry, F.; Nougairede, A.; Gould, E.A.; de Lamballerie, X. Flavivirus reverse genetic systems, construction techniques and applications: A historical perspective. Antiviral Res. 2015, 114, 67–85. [Google Scholar] [CrossRef] [PubMed]
- Shizuya, H.; Birren, B.; Kim, U.J.; Mancino, V.; Slepak, T.; Tachiiri, Y.; Simon, M. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl. Acad. Sci. USA 1992, 89, 8794–8797. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Widman, D.G.; Young, E.; Yount, B.L.; Plante, K.S.; Gallichotte, E.N.; Carbaugh, D.L.; Peck, K.M.; Plante, J.; Swanstrom, J.; Heise, M.T.; et al. A reverse genetics platform that spans the Zika virus family tree. mBio 2017, 8, e02014-16. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Kenney, H.; Chen, R.; Liu, G.; Manukyan, H.; Whitehead, S.S.; Laassri, M.; Chumakov, K.; Pletnev, A.G. A full-length infectious cDNA clone of Zika virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. mBio 2016, 7, e01114-16. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef] [PubMed]
- Setoh, Y.X.; Prow, N.A.; Peng, N.; Hugo, L.E.; Devine, G.; Hazlewood, J.E.; Suhrbier, A.; Khromykh, A.A. De Novo generation and characterization of new Zika virus isolate using sequence data from a microcephaly case. mSphere 2017, 2, e00190-17. [Google Scholar] [CrossRef] [PubMed]
- Atieh, T.; Baronti, C.; de Lamballerie, X.; Nougairede, A. Simple reverse genetics systems for Asian and African Zika viruses. Sci. Rep. 2016, 6, 39384. [Google Scholar] [CrossRef] [PubMed]
- Wichgers Schreur, P.J.; van Keulen, L.; Anjema, D.; Kant, J.; Kortekaas, J. Microencephaly in fetal piglets following in utero inoculation of Zika virus. Emerg. Microbes Infect. 2018, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Darbellay, J.; Lai, K.; Babiuk, S.; Berhane, Y.; Ambagala, A.; Wheler, C.; Wilson, D.; Walker, S.; Potter, A.; Gilmour, M.; et al. Neonatal pigs are susceptible to experimental Zika virus infection. Emerg. Microbes Infect. 2017, 6, e6. [Google Scholar] [CrossRef] [PubMed]
- Ragan, I.K.; Blizzard, E.L.; Gordy, P.; Bowen, R.A. Investigating the potential role of North American animals as hosts for Zika virus. Vector Borne Zoonotic Dis. 2017, 17, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Lecot, S.; Belouzard, S.; Dubuisson, J.; Rouille, Y. Bovine viral diarrhea virus entry is dependent on clathrin-mediated endocytosis. J. Virol. 2005, 79, 10826–10829. [Google Scholar] [CrossRef] [PubMed]
- Cureton, D.K.; Massol, R.H.; Whelan, S.P.; Kirchhausen, T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010, 6, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A mouse model of Zika virus pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A susceptible mouse model for Zika virus infection. PLoS Negl. Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Hollidge, B.; Daye, S.; Zeng, X.; Blancett, C.; Kuszpit, K.; Bocan, T.; Koehler, J.W.; Coyne, S.; Minogue, T.; et al. Neuropathogenesis of Zika virus in a highly susceptible immunocompetent mouse model after antibody blockade of type I interferon. PLoS Negl. Trop. Dis. 2017, 11, e0005296. [Google Scholar] [CrossRef] [PubMed]
- Larocca, R.A.; Abbink, P.; Peron, J.P.; Zanotto, P.M.; Iampietro, M.J.; Badamchi-Zadeh, A.; Boyd, M.; Ng’ang’a, D.; Kirilova, M.; Nityanandam, R.; et al. Vaccine protection against Zika virus from Brazil. Nature 2016, 536, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Manangeeswaran, M.; Ireland, D.D.; Verthelyi, D. Zika (PRVABC59) infection is associated with T cell infiltration and neurodegeneration in CNS of immunocompetent neonatal C57BL/6 mice. PLoS Pathog. 2016, 12, e1006004. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, N.C.; Nogueira, J.S.; Ressio, R.A.; Cirqueira, C.S.; Kimura, L.M.; Fernandes, K.R.; Cunha, M.S.; Souza, R.P.; Guerra, J.M. Experimental Zika virus infection induces spinal cord injury and encephalitis in newborn Swiss mice. Exp. Toxicol. Pathol. 2017, 69, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Zhang, A.J.; Chan, C.C.; Yip, C.C.; Mak, W.W.; Zhu, H.; Poon, V.K.; Tee, K.M.; Zhu, Z.; Cai, J.P.; et al. Zika virus infection in dexamethasone-immunosuppressed mice demonstrating disseminated infection with multi-organ involvement including orchitis effectively treated by recombinant type I interferons. EBioMedicine 2016, 14, 112–122. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Cell | Tissue | Growth Medium a | Culture Condition | Source (Catalog Number) b |
|---|---|---|---|---|---|
| Human | HEK | Embryo, kidney | MEM supplemented with 10% FBS, 2 mM L-glutamine, 0.1 mM NEAA, 1.0 mM SP, and PS | 37oC, 5% CO2 | ATCC (CRL-1573) |
| Human | Huh-7 | Liver | DMEM supplemented with 10% FBS, 0.1 mM NEAA, and PS | 37oC, 5% CO2 | Charles M. Rice, RU |
| Human | SH-SY5Y | Bone marrow | A 1:1 mixture of MEM and Ham's F-12 nutrient medium supplemented with 10% FBS, 0.1 mM NEAA, and PS | 37oC, 5% CO2 | ATCC (CRL-2266) |
| Mouse | MEF | Embryo (C57BL/6), fibroblast | DMEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | ATCC (SCRC-1008) |
| Mouse | NIH/3T3 | Embryo (NIH/Swiss), fibroblast | DMEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | ATCC (CRL-1658) |
| Mouse | NSC-34 | Motor neuron-like hybrid | DMEM (without SP) supplemented with 10% FBS and PS | 37oC, 5% CO2 | Cedarlane (CLU140) |
| Monkey | Vero | Kidney | α-MEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | ATCC (WHO-Vero) |
| Cow | BT | Turbinate | DMEM (without SP) supplemented with 10% HS and PS | 37oC, 5% CO2 | ATCC (CRL-1390) |
| Cow | MDBK | Kidney | DMEM (without SP) supplemented with 10% HS and PS | 37oC, 5% CO2 | ATCC (CCL-22) |
| Pig | ST | Testis | α-MEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | ATCC (CRL-1746) |
| Sheep | SFF-6 | Fetus, fibroblast | DMEM supplemented with 15% FBS and PS | 37oC, 5% CO2 | Irina A. Polejaeva, USU |
| Goat | GFF-4 | Fetus, fibroblast | DMEM supplemented with 15% FBS and PS | 37oC, 5% CO2 | Irina A. Polejaeva, USU |
| Horse | NBL-6 | Skin, dermis | EMEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | ATCC (CCL-57) |
| Dog | MDCK | Kidney | MEM supplemented with EBSS, 10% FBS, 0.1 mM NEAA, 1.0 mM SP, and PS | 37oC, 5% CO2 | ATCC (CCL-34) |
| Cat | CRFK | Kidney, cortex | MEM supplemented with EBSS, 10% HS, 0.1 mM NEAA, 1.0 mM SP, and PS | 37oC, 5% CO2 | ATCC (CCL-94) |
| Chicken | CEF | Embryo, fibroblast | DMEM supplemented with 10% FBS and PS | 37oC, 5% CO2 | Sung-June Byun, KNIAS |
| Mosquito | C6/36 | Larva (Aedes albopictus) | MEM supplemented with EBSS, 10% FBS, 2 mM L-glutamine, 0.1 mM NEAA, 1.0 mM SP, and PS | 28oC, 5% CO2 | ATCC (CRL-1660) |
| Oligonucleotide | Sequence a (5’ to 3’) | Position b | Direction |
|---|---|---|---|
| Z1RT | GCTATTGGGTTCATGCCACAGATGGTCATCA | 4531–4561 | Reverse |
| Z1F | tatgtttaaacAGTTGTTGATCTGTGTGAATCAGACTGCGA | 1–30 | Forward |
| Z1R | tatggcgcgccAGGACCACCTTGAGTATGATCTCTCTCATG | 4502–4531 | Reverse |
| Z2RT | ATTGTCATTGTGTCAATGTCAGTCACCACTA | 7369–7399 | Reverse |
| Z2F | tatgtttaaacTCATTGTTTGGAGGAATGTCCTGGTTCTCA | 2340–2369 | Forward |
| Z2R | tatggcgcgccTCAATGTCAGTCACCACTATTCCATCCACA | 7358–7387 | Reverse |
| Z3RT | CTCCAGTTCAGGCCCCAGATTGAAGGGTGGGG | 10603–10634 | Reverse |
| Z3F | tatgtttaaacGGAAGTCCCAGAGAGAGCCTGGAGCTCAGG | 5627–5656 | Forward |
| Z3R | tatggcgcgccAAGGGTGGGGAAGGTCGCCACCTTCTTTTC | 10583–10612 | Reverse |
| S123-5sp1F | ctaggatccttaattaacctgcagggggctgtta | Forward | |
| S123-5sp1R | GATCAACAACTctatagtgtcccctaaatc | 1–11 | Reverse |
| S1-5sp2F | ggacactatagAGTTGTTGATCTGTGTGAGTC | 1–21 | Forward |
| S1-5sp2R | tatccgcggTAGCGCAAACCCGGGGTTCCTGAAT | 860–884 | Reverse |
| S1-3roF | tatccgcggGGAAAAAGGGAGGACTTATGGTGTG | 10191–10215 | Forward |
| S1-3roR | agggcggccgcgtatgtcgcgttccgtacgttctagAGAAACCATGGATTTCCCCACACC | 10785–10807 | Reverse |
| S23-5sp2F | ggacactatagAGTTGTTGATCTGTGTGAATC | 1–21 | Forward |
| S23-5sp2R | tatccgcggAACGCAAAGCCAGGGTTCCTGAATA | 859–883 | Reverse |
| S23-3roF | tatccgcggGGGAAAAAGGGAAGACTTATGGTGT | 10190–10214 | Forward |
| S23-3roR | agggcggccgcgtatgtcgccttccgtacgttctagAGACCCATGGATTTCCCCACACCG | 10784–10807 | Reverse |
| ZikaC-F | tttgaattcGGTCTCATCAATAGATGGGGT | 297–317 | Forward |
| ZikaC-R | tttctcgagctattaTCGTCTCTTCTTCTCCTTCCT | 399–419 | Reverse |
| ZikaM-F | tttgaattcGCTGTGACGCTCCCCTCCCAT | 753–773 | Forward |
| ZikaM-R | tttctcgagctattaGACTCTAATCAAGTGCTTTGT | 828–848 | Reverse |
| ZikaE-F | tttgaattcCAGCACAGTGGGATGATCGTT | 1416–1436 | Forward |
| ZikaE-R | tttctcgagctattaTCCTAGGCTTCCAAAACCCCC | 1518–1538 | Reverse |
| ZikaNS4A-F | tttgaattcGGAGCGGCTTTTGGAGTGATG | 6465–6485 | Forward |
| ZikaNS4A-R | tttctcgagctattaGGTCTCCGGCAATTGGGCCGC | 6597–6617 | Reverse |
| ZikaNS4B-F | tttgaattcGTGACTGACATTGACACAATG | 7374–7394 | Forward |
| ZikaNS4B-R | tttctcgagctattaGGAAGTTGCGGCTGTGATCAG | 7506–7526 | Reverse |
| ZikaF | GAAGTGGAAGTCCCAGAGAG | 5622–5641 | Forward |
| ZikaR | TGCTGAGCTGTATGACCCG | 5757–5775 | Reverse |
| ZikaProbe | FAM-TGGAGCTCAGGCTTTGATTGGGTGAC-BHQ1 | 5646–5671 | Forward |
| VeroF | AGCGGGAAATCGTGCGTGAC | 624–643 | Forward |
| VeroR | CAATGGTGATGACCTGGCCA | 742–761 | Reverse |
| VeroProbe | HEX-CACGGCGGCTTCTAGCTCCTCCC-BHQ2 | 694–716 | Forward |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, S.-I.; Song, B.-H.; Frank, J.C.; Julander, J.G.; Olsen, A.L.; Polejaeva, I.A.; Davies, C.J.; White, K.L.; Lee, Y.-M. Functional Genomics and Immunologic Tools: The Impact of Viral and Host Genetic Variations on the Outcome of Zika Virus Infection. Viruses 2018, 10, 422. https://doi.org/10.3390/v10080422
Yun S-I, Song B-H, Frank JC, Julander JG, Olsen AL, Polejaeva IA, Davies CJ, White KL, Lee Y-M. Functional Genomics and Immunologic Tools: The Impact of Viral and Host Genetic Variations on the Outcome of Zika Virus Infection. Viruses. 2018; 10(8):422. https://doi.org/10.3390/v10080422
Chicago/Turabian StyleYun, Sang-Im, Byung-Hak Song, Jordan C. Frank, Justin G. Julander, Aaron L. Olsen, Irina A. Polejaeva, Christopher J. Davies, Kenneth L. White, and Young-Min Lee. 2018. "Functional Genomics and Immunologic Tools: The Impact of Viral and Host Genetic Variations on the Outcome of Zika Virus Infection" Viruses 10, no. 8: 422. https://doi.org/10.3390/v10080422
APA StyleYun, S.-I., Song, B.-H., Frank, J. C., Julander, J. G., Olsen, A. L., Polejaeva, I. A., Davies, C. J., White, K. L., & Lee, Y.-M. (2018). Functional Genomics and Immunologic Tools: The Impact of Viral and Host Genetic Variations on the Outcome of Zika Virus Infection. Viruses, 10(8), 422. https://doi.org/10.3390/v10080422