Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection

 , and
, and 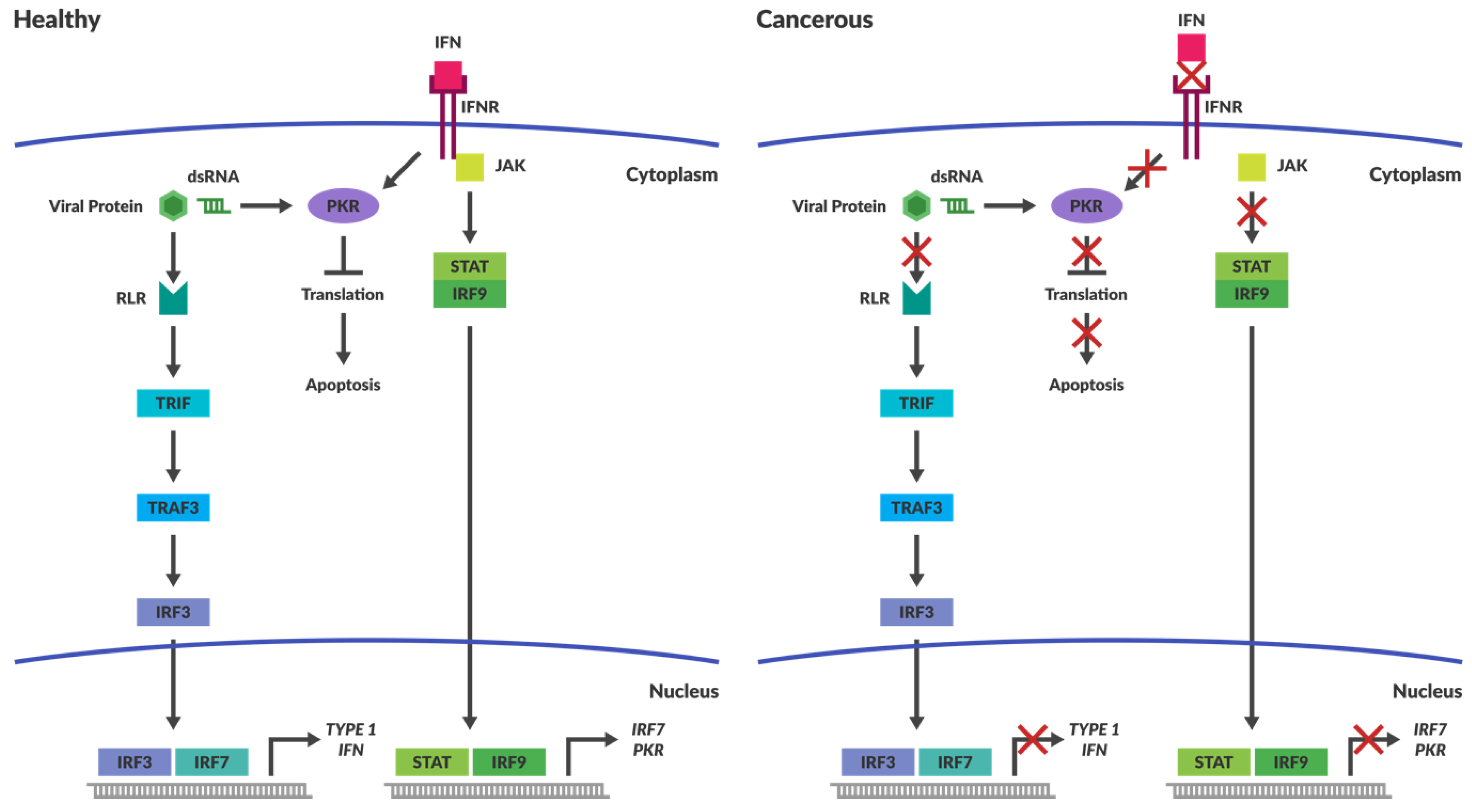{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Oncolytic Viruses
1.2. Fundamental Biology Directs Viral Engineering and Combination Therapy
1.3. Reovirus
2. Delivery
2.1. Barriers Preventing Infection
2.2. Circumventing Immune Clearance
3. Receptor Attachment and Entry
3.1. Receptor Mediated Selectivity
3.2. Viral Entry and Uncoating
4. Viral Replication and Appropriation of Host Intracellular Machinery
4.1. Intracellular Immune Surveillance
4.2. Cancers Have Compromised Immune Responses
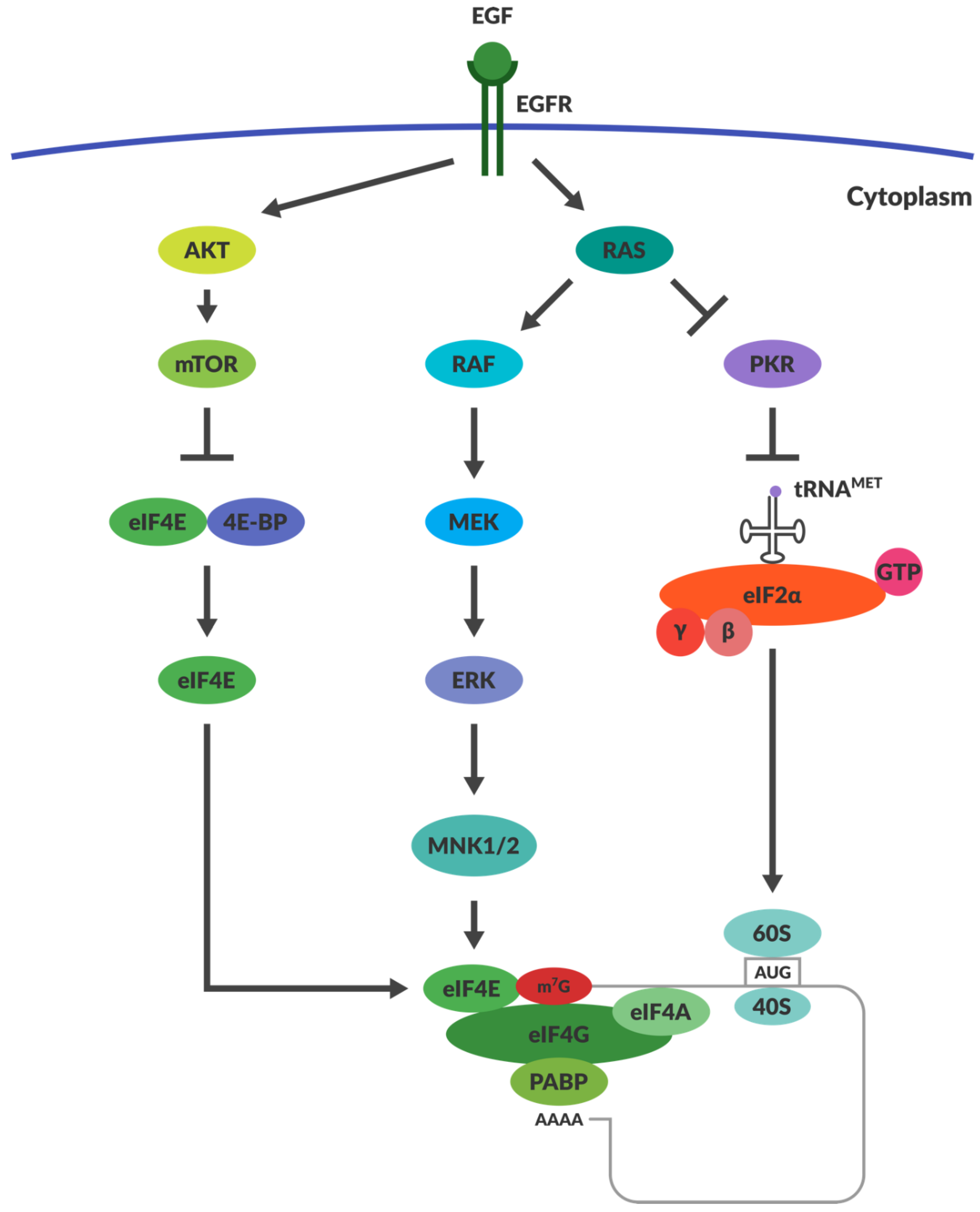
4.3. OVs Take Advantage of the Aberrant Signalling in Cancers
4.4. Does RAS Activation and PKR Inactivation Determine Reoviral Susceptibility?
4.5. RAS Activation May Not Be Responsible for Reoviral Tropism
4.6. Alternative Proposed Intracellular Mechanisms Governing Reoviral Tropism
5. Apoptosis Induction
6. Effects of the Tumour Microenvironment
6.1. Development of Cancer Immunotherapies
6.2. Debate Around the Importance of OV Immune Stimulation
6.3. Tumour Microenvironment and Reoviral Dissemination
7. Discussion
Funding
Conflicts of Interest
References
- Bierman, H.R.; Crile, D.M.; Dod, K.S.; Kelly, K.H.; Petrakis, N.L.; White, L.P.; Shimkin, M.B. Remissions in leukemia of childhood following acute infectious disease: Staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer 1953, 6, 591–605. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Berkrot, B. FDA Approves Amgen’s Injected Immunotherapy for Melanoma. Available online: http://www.reuters.com/article/2015/10/27/us-amgen-fda-idUSKCN0SL2YH20151027 (accessed on 9 November 2015).
- Anon. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Status from the Ema for Pancreatic Cancer. Available online: http://www.oncolyticsbiotech.com/news/press-release-details/2015/Oncolytics-Biotech-Inc-Announces-Receipt-of-Orphan-Drug-Status-from-the-EMA-for-Pancreatic-Cancer/default.aspx (accessed on 9 October 2015).
- Anon. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Designation from the U.S. FDA for Gastric Cancer. Available online: http://www.oncolyticsbiotech.com/news/press-release-details/2015/Oncolytics-Biotech-Inc-Announces-Receipt-of-Orphan-Drug-Designation-from-the-US-FDA-for-Gastric-Cancer/default.aspx (accessed on 9 October 2015).
- Anon. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Status from the EMA for Pancreatic Cancer. Available online: https://www.oncolyticsbiotech.com/press-releases/detail/348/oncolytics-biotech-inc-announces-receipt-of-orphan-drug (accessed on 30 July 2018).
- Anon. Oncolytics Biotech® Inc. Announces Receipt of Orphan Drug Status from the EMA for Ovarian Cancer. Available online: https://www.oncolyticsbiotech.com/press-releases/detail/340/oncolytics-biotech-inc-announces-receipt-of-orphan-drug (accessed on 30 July 2018).
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Yaacov, B.; Shiloach, T.; Eliahoo, E.; Kadouri, L.; Lotem, M.; Perlman, R.; Zakay-Rones, Z.; Panet, A.; Ben-Yehuda, D. The oncolytic activity of newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic immunotherapy: Dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [PubMed]
- Tapia, K.; Kim, W.-K.; Sun, Y.; Mercado-López, X.; Dunay, E.; Wise, M.; Adu, M.; López, C.B. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; de Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Hall, G.; Pulido, J.; Diaz, R.M.; Thompson, J.; Chong, H.; Selby, P.; Coffey, M.; Pandha, H.; Chester, J.; et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J. Clin. Investig. 2010, 120, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Puhlmann, M.; Brown, C.K.; Gnant, M.; Huang, J.; Libutti, S.K.; Alexander, H.R.; Bartlett, D.L. Vaccinia as a vector for tumor-directed Gene Therapy: Biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000, 7, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hikichi, M.; Kidokoro, M.; Haraguchi, T.; Iba, H.; Shida, H.; Tahara, H.; Nakamura, T. Microrna regulation of glycoprotein B5R in oncolytic vaccinia virus reduces viral pathogenicity without impairing its antitumor efficacy. Mol. Ther. 2011, 19, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Tedcastle, A.; Illingworth, S.; Brown, A.; Seymour, L.W.; Fisher, K.D. Actin-resistant dnase I expression from oncolytic adenovirus enadenotucirev enhances its intratumoral spread and reduces tumor growth. Mol. Ther. 2016, 24, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Antar, A.A.R.; Boehme, K.W.; Danthi, P.; Eby, E.A.; Guglielmi, K.M.; Holm, G.H.; Johnson, E.M.; Maginnis, M.S.; Naik, S.; et al. A plasmid-based reverse genetics system for animal double-stranded rna viruses. Cell Host Microbe 2007, 1, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Eaton, H.E.; Kobayashi, T.; Dermody, T.S.; Johnston, R.N.; Jais, P.H.; Shmulevitz, M. African swine fever virus NP868R capping enzyme promotes reovirus rescue during reverse genetics by promoting reovirus protein expression, virion assembly, and RNA incorporation into infectious virions. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Dautzenberg, I.J.; van den Wollenberg, D.J.; van den Hengel, S.K.; Limpens, R.W.; Barcena, M.; Koster, A.J.; Hoeben, R.C. Mammalian orthoreovirus T3D infects U-118 MG cell spheroids independent of junction adhesion molecule-a. Gene Ther. 2014, 21, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Garant, K.A.; zur Nieden, N.I.; Alain, T.; Loken, S.D.; Urbanski, S.J.; Forsyth, P.A.; Rancourt, D.E.; Lee, P.W.; Johnston, R.N. Attenuated reovirus displays oncolysis with reduced host toxicity. Br. J. Cancer 2011, 104, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Gujar, S.A.; Ahn, D.G.; Mohamed, A.; Lee, P.W. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 2012, 86, 7403–7413. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Johnston, R.N.; Shmulevitz, M. Potential for improving potency and specificity of reovirus oncolysis with next-generation reovirus variants. Viruses 2015, 7, 6251–6278. [Google Scholar] [CrossRef] [PubMed]
- Howells, A.; Marelli, G.; Lemoine, N.R.; Wang, Y. Oncolytic viruses-interaction of virus and tumor cells in the battle to eliminate cancer. Front. Oncol. 2017, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.T.; Liu, J.; McFadden, G. Bugs and drugs: Oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1817–1833. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Davydova, J.; Curiel, D.T.; Yamamoto, M. Combination of conditionally replicative adenovirus and standard chemotherapies shows synergistic antitumor effect in pancreatic cancer. Cancer Sci. 2009, 100, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L. Serologic grouping of reoviruses by hemagglutination-inhibition. Am. J. Hyg. 1960, 71, 242–249. [Google Scholar] [PubMed]
- Sabin, A.B. Reoviruses. A new group of respiratory and enteric viruses formerly classified as echo type 10 is described. Science 1959, 130, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Sokol, R.J.; Oberhaus, S.M.; Le, M.; Karrer, F.M.; Narkewicz, M.R.; Tyson, R.W.; Murphy, J.R.; Low, R.; Brown, W.R. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology 1998, 27, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Loken, S.D.; Norman, K.; Hirasawa, K.; Nodwell, M.; Lester, W.M.; Demetrick, D.J. Morbidity in immunosuppressed (SCID/NOD) mice treated with reovirus (dearing 3) as an anti-cancer biotherapeutic. Cancer Biol. Ther. 2004, 3, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, A.J.; Sipe, J.D.; Loh, P. Separation of ten reovirus genome segments by polyacrylamide gel electrophoresis. J. Virol. 1968, 2, 986–991. [Google Scholar] [PubMed]
- Mainou, B.A. The orchestra of reovirus cell entry. Curr. Clin. Microbiol. Rep. 2017, 4, 142–149. [Google Scholar] [CrossRef]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Attoui, H.; Biagini, P.; Stirling, J.; Mertens, P.P.C.; Cantaloube, J.-F.; Meyer, A.; de Micco, P.; de Lamballerie, X. Sequence characterization of ndelle virus genome segments 1, 5, 7, 8, and 10: Evidence for reassignment to the genus orthoreovirus, family reoviridae. Biochem. Biophys. Res. Commun. 2001, 287, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.R.; Stanish, S.M.; Cox, D.C. Differential sensitivity of normal and transformed human cells to reovirus infection. J. Virol. 1978, 28, 444–449. [Google Scholar] [PubMed]
- Hashiro, G.; Loh, P.C.; Yau, J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977, 54, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Sachdev, E.; Mita, A.C.; Mita, M.M. Clinical development of reovirus for cancer therapy: An oncolytic virus with immune-mediated antitumor activity. World J. Methodol. 2016, 6, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Minuk, G.Y.; Paul, R.W.; Lee, P.W. The prevalence of antibodies to reovirus type 3 in adults with idiopathic cholestatic liver disease. J. Med. Virol. 1985, 16, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Tai, J.H.; Williams, J.V.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr.; Dermody, T.S. Prevalence of reovirus-specific antibodies in young children in nashville, tennessee. J. Infect. Dis. 2005, 191, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Leers, W.D.; Rozee, K.R. A survey of reovirus antibodies in sera of urban children. Can. Med. Assoc. J. 1966, 94, 1040–1042. [Google Scholar] [PubMed]
- White, C.L.; Twigger, K.R.; Vidal, L.; De Bono, J.S.; Coffey, M.; Heinemann, L.; Morgan, R.; Merrick, A.; Errington, F.; Vile, R.G.; et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (dearing type 3) during a phase I clinical trial. Gene Ther. 2008, 15, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Inoue, S.; Kaminade, T.; Hotani, T.; Katayama, Y.; Hosoyamada, E.; Terasawa, Y.; Tachibana, M.; Mizuguchi, H. Cationic liposome-mediated delivery of reovirus enhances the tumor cell-killing efficiencies of reovirus in reovirus-resistant tumor cells. Int. J. Pharm. 2017, 524, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.J.; Prestwich, R.J.; Kottke, T.; Errington, F.; Thompson, J.M.; Harrington, K.J.; Pandha, H.S.; Coffey, M.; Selby, P.J.; Vile, R.G.; et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther. 2009, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Adair, R.A.; Roulstone, V.; Scott, K.J.; Morgan, R.; Nuovo, G.J.; Fuller, M.; Beirne, D.; West, E.J.; Jennings, V.A.; Rose, A.; et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci. Transl. Med. 2012, 4, 138ra177. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hall, R.R.; Lesniak, M.S.; Ahmed, A.U. Stem cell-based cell carrier for targeted oncolytic virotherapy: Translational opportunity and open questions. Viruses 2015, 7, 6200–6217. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, M. Reovirus safety study for proliferation and differentiation of human adipose-derived mesenchymal stem cells. J. Microbiol. 2017, 55, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, Y.; Hotani, T.; Katayama, Y.; Tachibana, M.; Mizuguchi, H.; Sakurai, F. Activity levels of cathepsins B and L in tumor cells are a biomarker for efficacy of reovirus-mediated tumor cell killing. Cancer Gene Ther. 2015, 22, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Dorig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Yu, Z.; Chan, M.K.; O-charoenrat, P.; Eisenberg, D.P.; Shah, J.P.; Singh, B.; Fong, Y.; Wong, R.J. Enhanced nectin-1 expression and herpes oncolytic sensitivity in highly migratory and invasive carcinoma. Clin. Cancer Res. 2005, 11, 4889–4897. [Google Scholar] [CrossRef] [PubMed]
- Hulin-Curtis, S.L.; Uusi-Kerttula, H.; Jones, R.; Hanna, L.; Chester, J.D.; Parker, A.L. Evaluation of CD46 re-targeted adenoviral vectors for clinical ovarian cancer intraperitoneal therapy. Cancer Gene Ther. 2016, 23, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Gentsch, J.R.; Pacitti, A.F. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology 1987, 161, 245–248. [Google Scholar] [CrossRef]
- Reiter, D.M.; Frierson, J.M.; Halvorson, E.E.; Kobayashi, T.; Dermody, T.S.; Stehle, T. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog. 2011, 7, e1002166. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Stencel, J.E.; Liu, Y.; Blaum, B.S.; Reiter, D.M.; Feizi, T.; Dermody, T.S.; Stehle, T. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog. 2012, 8, e1003078. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P.; Holm, G.H.; Stehle, T.; Dermody, T.S. Reovirus receptors, cell entry, and proapoptotic signaling. In Viral Entry into Host Cells; Springer: New York, NY, USA, 2013; pp. 42–71. [Google Scholar]
- Barton, E.S.; Forrest, J.C.; Connolly, J.L.; Chappell, J.D.; Liu, Y.; Schnell, F.J.; Nusrat, A.; Parkos, C.A.; Dermody, T.S. Junction adhesion molecule is a receptor for reovirus. Cell 2001, 104, 441–451. [Google Scholar] [CrossRef]
- Lai, C.M.; Mainou, B.A.; Kim, K.S.; Dermody, T.S. Directional release of reovirus from the apical surface of polarized endothelial cells. MBio 2013, 4, e00049-13. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S.; Forrest, J.C.; Kopecky-Bromberg, S.A.; Dickeson, S.K.; Santoro, S.A.; Zutter, M.M.; Nemerow, G.R.; Bergelson, J.M.; Dermody, T.S. β1 integrin mediates internalization of mammalian reovirus. J. Virol. 2006, 80, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Sturzenbecker, L.J.; Nibert, M.; Furlong, D.; Fields, B.N. Intracellular digestion of reovirus particles requires a low PH and is an essential step in the viral infectious cycle. J. Virol. 1987, 61, 2351–2361. [Google Scholar] [PubMed]
- Ebert, D.H.; Deussing, J.; Peters, C.; Dermody, T.S. Cathepsin l and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002, 277, 24609–24617. [Google Scholar] [CrossRef] [PubMed]
- Madren, J.A.; Sarkar, P.; Danthi, P. Cell entry-associated conformational changes in reovirus particles are controlled by host protease activity. J. Virol. 2012, 86, 3466–3473. [Google Scholar] [CrossRef] [PubMed]
- Alain, T.; Kim, T.S.; Lun, X.; Liacini, A.; Schiff, L.A.; Senger, D.L.; Forsyth, P.A. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol. Ther. 2007, 15, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Pan, L.Z.; Garant, K.; Pan, D.; Lee, P.W. Oncogenic ras promotes reovirus spread by suppressing ifn-beta production through negative regulation of rig-I signaling. Cancer Res. 2010, 70, 4912–4921. [Google Scholar] [CrossRef] [PubMed]
- Amerongen, H.M.; Wilson, G.A.; Fields, B.N.; Neutra, M.R. Proteolytic processing of reovirus is required for adherence to intestinal m cells. J. Virol. 1994, 68, 8428–8432. [Google Scholar] [PubMed]
- Boulant, S.; Stanifer, M.; Kural, C.; Cureton, D.K.; Massol, R.; Nibert, M.L.; Kirchhausen, T. Similar uptake but different trafficking and escape routes of reovirus virions and infectious subvirion particles imaged in polarized madin-darby canine kidney cells. Mol. Biol Cell 2013, 24, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Barton, E.S.; Youree, B.E.; Ebert, D.H.; Forrest, J.C.; Connolly, J.L.; Valyi-Nagy, T.; Washington, K.; Wetzel, J.D.; Dermody, T.S. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J. Clin. Investig. 2003, 111, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Van den Wollenberg, D.J.M.; Dautzenberg, I.J.C.; van den Hengel, S.K.; Cramer, S.J.; de Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-a. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [PubMed]
- Bull, C.; Stoel, M.A.; den Brok, M.H.; Adema, G.J. Sialic acids sweeten a tumor’s life. Cancer Res. 2014, 74, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Tang, D.; Lee, P.W. Evidence that the epidermal growth factor receptor on host cells confers reovirus infection efficiency. Virology 1993, 197, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Lee, P.W. The v-erbb oncogene confers enhanced cellular susceptibility to reovirus infection. J. Virol. 1996, 70, 612–616. [Google Scholar] [PubMed]
- Clemens, M.J.; Pain, V.M.; Wong, S.T.; Henshaw, E.C. Phosphorylation inhibits guanine nucleotide exchange on eukaryotic initiation factor 2. Nature 1982, 296, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, T.; Pavitt, G.D.; Zhang, F.; Dever, T.E.; Hinnebusch, A.G. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (EIF2α) to the regulatory subunits of guanine nucleotide exchange factor EIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 2001, 21, 5018–5030. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Imani, F.; Jacobs, B.L. Inhibitory activity for the interferon-induced protein kinase is associated with the reovirus serotype 1 sigma 3 protein. Proc. Natl. Acad. Sci. USA 1988, 85, 7887–7891. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.D.; Holm, G.H.; Boehme, K.W. Differential delivery of genomic dsrna causes reovirus strain-specific differences in IRF3 activation. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zurney, J.; Kobayashi, T.; Holm, G.H.; Dermody, T.S.; Sherry, B. Reovirus µ2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J. Virol. 2009, 83, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a m(r) 90,000 phosphoprotein with protein kinase pkr in cells exhibiting enhanced phosphorylation of translation initiation factor EIF-2α and premature shutoff of protein synthesis after infection with γ 134.5- mutants of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1995, 92, 10516–10520. [Google Scholar] [PubMed]
- He, B.; Gross, M.; Roizman, B. The γ(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded rna-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [PubMed]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 US11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Martuza, R.L.; Rabkin, S.D. Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte–macrophage colony-stimulating factor. Mol. Ther. 2000, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Breitbach, C.J.; Le Boeuf, F.; Wang, J.; Storbeck, C.; Ilkow, C.; Diallo, J.S.; Falls, T.; Burns, J.; Garcia, V.; et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 2012, 20, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the RAS signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting ras signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Hirasawa, K.; Yang, A.-D.; Shields, M.A.; Lee, P.W.K. Reovirus oncolysis: The RAS/RALGEF/P38 pathway dictates host cell permissiveness to reovirus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 11099–11104. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; van den Wollenberg, D.J.; Borel Rinkes, I.H.; Hoeben, R.C.; Kranenburg, O. Sensitization to apoptosis underlies krasd12-dependent oncolysis of murine C26 colorectal carcinoma cells by reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [PubMed]
- Smakman, N.; van den Wollenberg, D.J.; Elias, S.G.; Sasazuki, T.; Shirasawa, S.; Hoeben, R.C.; Borel Rinkes, I.H.; Kranenburg, O. KRAS(D13) promotes apoptosis of human colorectal tumor cells by reovirust3d and oxaliplatin but not by tumor necrosis factor-related apoptosis-inducing ligand. Cancer Res. 2006, 66, 5403–5408. [Google Scholar] [CrossRef] [PubMed]
- Van Houdt, W.J.; Smakman, N.; van den Wollenberg, D.J.; Emmink, B.L.; Veenendaal, L.M.; van Diest, P.J.; Hoeben, R.C.; Borel Rinkes, I.H.; Kranenburg, O. Transient infection of freshly isolated human colorectal tumor cells by reovirus T3D intermediate subviral particles. Cancer Gene Ther. 2008, 15, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Samuel, C.E. Protein kinase pkr plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 2007, 81, 8192–8200. [Google Scholar] [CrossRef] [PubMed]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the egfr pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Schmechel, S.C.; Williams, B.R.; Silverman, R.H.; Schiff, L.A. Involvement of the interferon-regulated antiviral proteins PKR and rnase L in reovirus-induced shutoff of cellular translation. J. Virol. 2005, 79, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Mainou, B.A.; Dermody, T.S. Transport to late endosomes is required for efficient reovirus infection. J. Virol. 2012, 86, 8346–8358. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Parker, J.S.; Ehrlich, M.; Kirchhausen, T.; Nibert, M.L. The delta region of outer-capsid protein micro 1 undergoes conformational change and release from reovirus particles during cell entry. J. Virol. 2003, 77, 13361–13375. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Chandran, K.; Baker, T.S.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus membrane-penetration protein, MU1, in a complex with is protector protein, sigma3. Cell 2002, 108, 283–295. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and p-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Carroll, K.; Hastings, C.; Miller, C.L. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of EIF2α phosphorylation and PKR. J. Virol. 2011, 85, 8798–8810. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Hastings, C.; Miller, C.L. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 2009, 83, 11090–11101. [Google Scholar] [CrossRef] [PubMed]
- Skup, D.; Millward, S. Reovirus-induced modification of CAP-dependent translation in infected L cells. Proc. Natl. Acad. Sci. USA 1980, 77, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.; Lemay, G.; Millward, S. The viral protein sigma 3 participates in translation of late viral mRNA in reovirus-infected L cells. J. Virol. 1987, 61, 2472–2479. [Google Scholar] [PubMed]
- Zamora, P.F.; Hu, L.; Knowlton, J.J.; Lahr, R.M.; Moreno, R.A.; Berman, A.J.; Prasad, B.V.V.; Dermody, T.S. Reovirus nonstructural protein sigmans acts as an RNA stability factor promoting viral genome replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Gingras, A.-C.; Donzé, O.; Lin, T.-A.; Lawrence, J.C.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-CAP function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Yu, S.L.; Chen, J.J.; Chang, S.Y.; Yan, B.S.; Hong, Q.S.; Singh, S.; Kao, C.L.; Chen, H.Y.; Su, K.Y.; et al. Enterovirus-induced MIR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor EIF4E. Cell Host Microbe 2011, 9, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis c virus RNA abundance by a liver-specific microrna. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded micrornas: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Garofalo, M.; Valeri, N.; Roulstone, V.; Volinia, S.; Cohn, D.E.; Phelps, M.; Harrington, K.J.; Vile, R.; Melcher, A.; et al. Reovirus-associated reduction of microRNA-let-7D is related to the increased apoptotic death of cancer cells in clinical samples. Mod. Pathol. 2012, 25, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, M.A.; Leber, M.F.; Bossow, S.; Singh, M.; Engeland, C.E.; Albert, J.; Grossardt, C.; Jager, D.; von Kalle, C.; Ungerechts, G. Microrna-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Ther. 2014, 21, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Kotzin, J.J.; Mowel, W.K.; Henao-Mejia, J. Viruses HIJACK a host lncRNA to replicate. Science 2017, 358, 993–994. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xu, J.; Wang, Y.; Cao, X. An interferon-independent lncrna promotes viral replication by modulating cellular metabolism. Science 2017, 358, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.L.; Squier, M.K.; Rodgers, S.E.; Schneider, B.E.; Oberhaus, S.M.; Grdina, T.A.; Cohen, J.J.; Dermody, T.S. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J. Virol. 1995, 69, 6972–6979. [Google Scholar] [PubMed]
- Rodgers, S.E.; Barton, E.S.; Oberhaus, S.M.; Pike, B.; Gibson, C.A.; Tyler, K.L.; Dermody, T.S. Reovirus-induced apoptosis of mdck cells is not linked to viral yield and is blocked by BCL-2. J. Virol. 1997, 71, 2540–2546. [Google Scholar] [PubMed]
- Boehme, K.W.; Guglielmi, K.M.; Dermody, T.S. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. USA 2009, 106, 19986–19991. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, C.C.; Richardson-Burns, S.M.; Goody, R.J.; Robinson, B.A.; Debiasi, R.L.; Tyler, K.L. Nonstructural protein sigma1s is a determinant of reovirus virulence and influences the kinetics and severity of apoptosis induction in the heart and central nervous system. J. Virol. 2005, 79, 2743–2753. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.W.; Hammer, K.; Tollefson, W.C.; Konopka-Anstadt, J.L.; Kobayashi, T.; Dermody, T.S. Nonstructural protein sigma1s mediates reovirus-induced cell cycle arrest and apoptosis. J. Virol. 2013, 87, 12967–12979. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Fields, B.N. A carboxy-terminal fragment of protein mu 1/MU 1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J. Virol. 1992, 66, 6408–6418. [Google Scholar] [PubMed]
- Coffey, C.M.; Sheh, A.; Kim, I.S.; Chandran, K.; Nibert, M.L.; Parker, J.S.L. Reovirus outer capsid protein μ1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J. Virol. 2006, 80, 8422–8438. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Meintzer, S.M.; Gibson, S.; Widmann, C.; Garrington, T.P.; Johnson, G.L.; Tyler, K.L. Reovirus-induced apoptosis is mediated by trail. J. Virol. 2000, 74, 8135–8139. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Meintzer, S.M.; Spalding, A.C.; Johnson, G.L.; Tyler, K.L. Caspase 8-dependent sensitization of cancer cells to trail-induced apoptosis following reovirus-infection. Oncogene 2001, 20, 6910–6919. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Bickel, R.J.; Tyler, K.L. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ. 2002, 9, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Kominsky, D.J.; Bickel, R.J.; Tyler, K.L. Reovirus-induced apoptosis requires mitochondrial release of smac/diablo and involves reduction of cellular inhibitor of apoptosis protein levels. J. Virol. 2002, 76, 11414–11424. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, J.J.; Dermody, T.S.; Holm, G.H. Apoptosis induced by mammalian reovirus is beta interferon (ifn) independent and enhanced by ifn regulatory factor 3-and NF-κb-dependent expression of noxa. J. Virol. 2012, 86, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Rodgers, S.E.; Clarke, P.; Ballard, D.W.; Kerr, L.D.; Tyler, K.L.; Dermody, T.S. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J. Virol. 2000, 74, 2981–2989. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Meintzer, S.M.; Wang, Y.; Moffitt, L.A.; Richardson-Burns, S.M.; Johnson, G.L.; Tyler, K.L. Jnk regulates the release of proapoptotic mitochondrial factors in reovirus-infected cells. J. Virol. 2004, 78, 13132–13138. [Google Scholar] [CrossRef] [PubMed]
- Garant, K.A.; Shmulevitz, M.; Pan, L.; Daigle, R.M.; Ahn, D.G.; Gujar, S.A.; Lee, P.W. Oncolytic reovirus induces intracellular redistribution of RAS to promote apoptosis and progeny virus release. Oncogene 2016, 35, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Holm, G.H.; Zurney, J.; Tumilasci, V.; Leveille, S.; Danthi, P.; Hiscott, J.; Sherry, B.; Dermody, T.S. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J. Biol. Chem. 2007, 282, 21953–21961. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-i and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, P.; Li, H.; Du, X.; Liu, M.; Huang, Q.; Wang, Y.; Wang, S. The efficacy of oncolytic adenovirus is mediated by T-cell responses against virus and tumor in syrian hamster model. Clin. Cancer Res. 2017, 23, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Coffey, M.C.; Thompson, B.G.; Yoon, C.S.; Waisman, D.M.; Lee, P.W. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003, 63, 348–353. [Google Scholar] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination therapy with reovirus and anti-PD-1 blockade controls tumor growth through innate and adaptive immune responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.; Kottke, T.; Thompson, J.; Rajani, K.; Zaidi, S.; Evgin, L.; Coffey, M.; Ralph, C.; Diaz, R.; Pandha, H.; et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumour therapy. Gene Ther. 2017, 24, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Tachibana, M.; Kurisu, N.; Oya, Y.; Terasawa, Y.; Goda, H.; Kobiyama, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H. Oncolytic reovirus inhibits immunosuppressive activity of myeloid-derived suppressor cells in a TLR3-dependent manner. J. Immunol. 2018, ji1700435. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.A.; Meyers, D.E.; Thirukkumaran, C.M.; Liu, P.J.; Gratton, K.; Spurrell, J.; Shi, Q.; Thakur, S.; Morris, D.G. Oncolytic reovirus and immune checkpoint inhibition as a novel immunotherapeutic strategy for breast cancer. Cancers 2018, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Lawson, K.A.; Mostafa, A.A.; Shi, Z.Q.; Spurrell, J.; Chen, W.; Kawakami, J.; Gratton, K.; Thakur, S.; Morris, D.G. Repurposing sunitinib with oncolytic reovirus as a novel immunotherapeutic strategy for renal cell carcinoma. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wu, K.; Visconte, V.; Anwer, F.; Calton, C.M.; Carew, J.S.; Nawrocki, S.T. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-l1 therapy. Leukemia 2018, 32, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Parakrama, R.; Chaudhary, I.; Coffey, M.C.; Goel, S.; Maitra, R. Immune response to reovirus (reo) in phase i study with chemotherapy in patients with kras mutant metastatic colorectal cancer (MCRC). Am. Soc. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Bodkin, D.K.; Nibert, M.L.; Fields, B.N. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J. Virol. 1989, 63, 4676–4681. [Google Scholar] [PubMed]
- Bass, D.M.; Bodkin, D.; Dambrauskas, R.; Trier, J.S.; Fields, B.N.; Wolf, J.L. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J. Virol. 1990, 64, 1830–1833. [Google Scholar] [PubMed]
- Figova, K.; Hrabeta, J.; Eckschlager, T. Anticancer efficiency of reovirus in normoxia and hypoxia. Folia Biol. 2013, 59, 68–75. [Google Scholar] [PubMed]
- Pipiya, T.; Sauthoff, H.; Huang, Y.Q.; Chang, B.; Cheng, J.; Heitner, S.; Chen, S.; Rom, W.N.; Hay, J.G. Hypoxia reduces adenoviral replication in cancer cells by downregulation of viral protein expression. Gene Ther. 2005, 12, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.I.; Watson, I.R.; Der, S.D.; Ohh, M. Loss of vhl confers hypoxia-inducible factor (HIF)-dependent resistance to vesicular stomatitis virus: Role of hif in antiviral response. J. Virol. 2006, 80, 10712–10723. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourhill, T.; Mori, Y.; Rancourt, D.E.; Shmulevitz, M.; Johnston, R.N. Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection. Viruses 2018, 10, 421. https://doi.org/10.3390/v10080421
Bourhill T, Mori Y, Rancourt DE, Shmulevitz M, Johnston RN. Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection. Viruses. 2018; 10(8):421. https://doi.org/10.3390/v10080421
Chicago/Turabian StyleBourhill, Tarryn, Yoshinori Mori, Derrick E. Rancourt, Maya Shmulevitz, and Randal N. Johnston. 2018. "Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection" Viruses 10, no. 8: 421. https://doi.org/10.3390/v10080421
APA StyleBourhill, T., Mori, Y., Rancourt, D. E., Shmulevitz, M., & Johnston, R. N. (2018). Going (Reo)Viral: Factors Promoting Successful Reoviral Oncolytic Infection. Viruses, 10(8), 421. https://doi.org/10.3390/v10080421