Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Guide RNAs (gRNAs) Design
2.2. Off-Target Analysis
2.3. Cloning gRNAs
2.4. Cells
2.5. DNA and RNA Extraction, Retrotranscription, and Real-Time (RT) PCR
2.6. Transfection
2.7. Western Blot Analysis
2.8. Statistics
3. Results
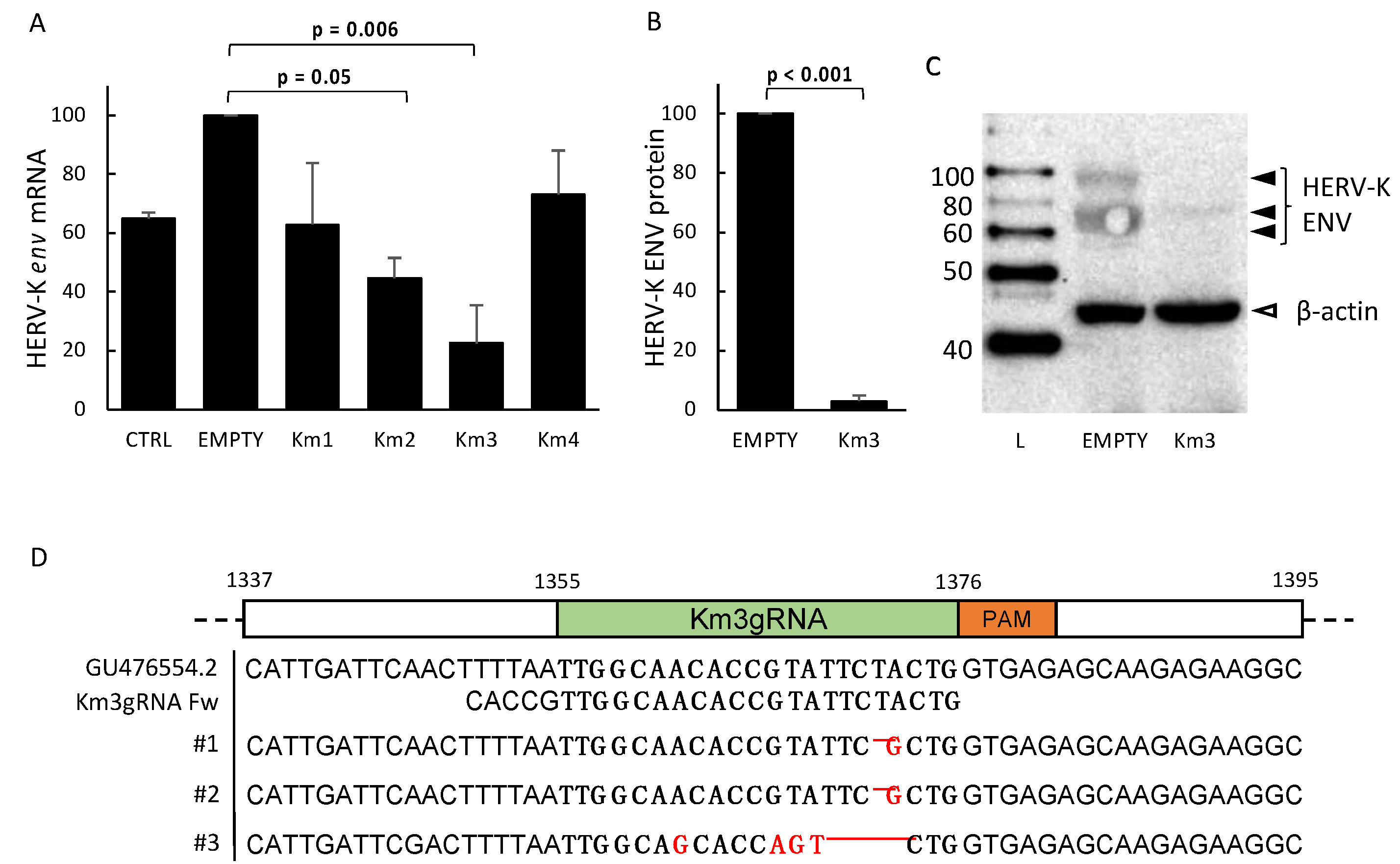
3.1. Setting and Validation of HERV-Kenv-Directed Guide RNAs
3.2. Effects of HERV-Kenv Gene Disruption on Molecules Central to Signaling Networks
3.3. HERV-Kenv Gene Disruption Affects SF2/ASF and TDP-43 Proteins
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Griffiths, D.J. Genome Biology. Genome Biol. 2001, 2, REVIEWS1017. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. ENCODE Project Writes Eulogy for Junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Downey, R.F.; Sullivan, F.J.; Wang-Johanning, F.; Ambs, S.; Giles, F.J.; Glynn, S.A. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int. J. Cancer 2014, 137, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Goering, W.; Schmitt, K.; Dostert, M.; Schaal, H.; Deenen, R.; Mayer, J.; Schulz, W.A. Human endogenous retrovirus HERV-K(HML-2) activity in prostate cancer is dominated by a few loci. Prostate 2015, 75, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Agoni, L.; Guha, C.; Lenz, J. Detection of Human Endogenous Retrovirus K (HERV-K) Transcripts in Human Prostate Cancer Cell Lines. Front. Oncol. 2013, 3, 180. [Google Scholar] [CrossRef] [PubMed]
- Carlini, F.; Ridolfi, B.; Molinari, A.; Parisi, C.; Bozzuto, G.; Toccacieli, L.; Formisano, G.; Orsi, D.D.; Paradisi, S.; Grober, O.M.V.; et al. The reverse transcription inhibitor Abacavir shows anticancer activity in prostate cancer cell lines. PLoS ONE 2010, 5, e14221. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Meng, Z.; Gan, Y.; Wang, X.; Xu, F.; Gu, Y.; Xu, X.; Tang, J.; Zhou, H.; Zhang, X.; et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating β-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia 2013, 27, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.; Sauter, M.; Schmitt, M.; Baumert, B.; Best, B.; Boese, A.; Roemer, K.; Mueller-Lantzsch, N. Human endogenous retrovirus protein Rec interacts with the testicular zinc-finger protein and androgen receptor. J. Gen. Virol. 2010, 91, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cao, M.; Iduma, P.; Karachaliou, N.; Santarpia, M.; Blanco, J.; Rosell, R. Human endogenous retroviruses and cancer. Cancer Biol. Med. 2016, 13, 483–488. [Google Scholar] [PubMed]
- Lemaître, C.; Tsang, J.; Bireau, C.; Heidmann, T.; Dewannieux, M. A human endogenous retrovirus-derived gene that can contribute to oncogenesis by activating the ERK pathway and inducing migration and invasion. PLoS Pathog. 2017, 13, e1006451. [Google Scholar] [CrossRef] [PubMed]
- Jacquenet, S.; Decimo, D.; Muriaux, D.; Darlix, J.L. Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology 2005, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Maslon, M.M.; Heras, S.R.; Bellora, N.; Eyras, E.; Cáceres, J.F. The translational landscape of the splicing factor SRSF1 and its role in mitosis. eLife 2014, 3, e02028. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; Stanchina, E.D.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhang, Z.; Sinha, R.; Karni, R.; Krainer, A.R. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol. 2010, 17, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Zhang, H.; Du, C.; Liu, X.; Zhu, S.; Zhang, W.; Li, Z.; Gao, C.; Zhao, X.; Mei, M.; et al. Correlation of SRSF1 and PRMT1 expression with clinical status of pediatric acute lymphoblastic leukemia. J. Hematol. Oncol. 2012, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Li, Y.; Ning, J.; Sun, D.; Lin, L.; Liu, X. HnRNP A1/A2 and SF2/ASF Regulate Alternative Splicing of Interferon Regulatory Factor-3 and Affect Immunomodulatory Functions in Human Non-Small Cell Lung Cancer Cells. PLoS ONE 2013, 8, e62729. [Google Scholar] [CrossRef] [PubMed]
- Shimoni-Sebag, A.; Lebenthal-Loinger, I.; Zender, L.; Karni, R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1-MAPK-ERK activation and cellular transformation. Carcinogenesis 2013, 34, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, T.; Zhang, X.; Wang, Q.; Zhang, J.; Ji, W.; Ma, Y. Splicing factor 2/alternative splicing factor contributes to extracellular signal-regulated kinase activation in hepatocellular carcinoma cells. Mol. Med. Rep. 2015, 12, 3890–3894. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Su, L.; Chen, X.; Li, P.; Cai, Q.; Yu, B.; Liu, B.; Wu, W.; Zhu, Z. MALAT1 promotes cell proliferation in gastric cancer by recruiting SF2/ASF. Biomed. Pharmacother. 2014, 68, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Q.; Ling, X.H.; Yuan, R.Q.; Chen, Z.Y.; Yang, S.B.; Huang, H.X.; Zhong, W.D.; Qiu, S.P. miR-30c suppresses prostate cancer survival by targeting the ASF/SF2 splicing factor oncoprotein. Mol. Med. Rep. 2017, 16, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Olshavsky, N.A.; Comstock, C.E.S.; Schiewer, M.J.; Augello, M.A.; Hyslop, T.; Sette, C.; Zhang, J.; Parysek, L.M.; Knudsen, K.E. Identification of ASF/SF2 as a Critical, Allele-Specific Effector of the Cyclin D1b Oncogene. Cancer Res. 2010, 70, 3975–3984. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2013, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chakrabartty, A. Phase to Phase with TDP-43. Biochemistry 2017, 56, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Cao, K.; Liu, R.; Huang, J.; Xia, K.; Tang, J.; Chen, X.; Zhou, M.; Xie, H.; Zhou, J. Identification of TDP-43 as an oncogene in melanoma and its function during melanoma pathogenesis. Cancer Biol. Ther. 2016, 18, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.Y.; Tan, O.; Liu, B.; Trahair, T.; Liu, T.; Haber, M.; Norris, M.D.; Marshall, G.M.; Cheung, B.B. High TDP43 expression is required for TRIM16-induced inhibition of cancer cell growth and correlated with good prognosis of neuroblastoma and breast cancer patients. Cancer Lett. 2016, 374, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fan, Z.; Mcgee, W.; Chen, M.; Kong, R.; Wen, P.; Xiao, T.; Chen, X.; Liu, J.; Zhu, L.; et al. TDP-43 regulates cancer-associated microRNAs. Protein Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wang, S.; Jia, W. Caspase independent cleavages of TDP-43 generates 35kD fragment that cause apoptosis of breast cancer cells. Biochem. Biophys. Res. Commun. 2018, 497, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Geldern, G.V.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Sedelaar, J.M.; Isaacs, J.T. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate 2009, 69, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Poddighe, L.; Astone, V.; Delogu, G.; Arru, G.; Sotgiu, S.; Serra, C.; Dolei, A. Novel reliable real-time PCR for differential detection of MSRVenv and syncytin-1 in RNA and DNA from patients with multiple sclerosis. J. Virol. Methods 2009, 161, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Uleri, E.; Mei, A.; Mameli, G.; Poddighe, L.; Serra, C.; Dolei, A. HIV Tat acts on endogenous retroviruses of the W family and this occurs via Toll-like receptor 4. AIDS 2014, 28, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Uleri, E.; Beltrami, S.; Gordon, J.; Dolei, A.; Sariyer, I.K. Extinction of Tumor Antigen Expression by SF2/ASF in JCV-Transformed Cells. Gen. Cancer 2011, 2, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [PubMed]
- Zhou, F.; Li, M.; Wei, Y.; Lin, K.; Lu, Y.; Shen, J.; Johanning, G.L.; Wang-Johanning, F. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget 2016, 7, 84093–84117. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nasu, Y.; Kashiwakura, Y.; Kusumi, N.; Tamayose, K.; Nagai, A.; Sasano, T.; Shimada, T.; Daida, H.; Kumon, H. Adeno-Associated Virus 2-Mediated Intratumoral Prostate Cancer Gene Therapy: Long-Term Maspin Expression Efficiently Suppresses Tumor Growth. Hum. Gene Ther. 2005, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Shorter, J. Biology and Pathobiology of TDP-43 and Emergent Therapeutic Strategies. CSH Perspect. Med. 2016, 7, A024554. [Google Scholar] [CrossRef] [PubMed]
- Rogers, G.L.; Cannon, P.M. Gene Therapy Approaches to Human Immunodeficiency Virus and Other Infectious Diseases. Hematol. Oncol. Clin. N. Am. 2017, 31, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Jubair, L.; Mcmillan, N.A. The Therapeutic Potential of CRISPR/Cas9 Systems in Oncogene-Addicted Cancer Types: Virally Driven Cancers as a Model System. Mol. Ther. Nucl. Acids 2017, 8, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lessard, S.; Francioli, L.; Alfoldi, J.; Tardif, J.C.; Ellinor, P.T.; Macarthur, D.G.; Lettre, G.; Orkin, S.H.; Canver, M.C. Human genetic variation alters CRISPR-Cas9 on- and off-targeting specificity at therapeutically implicated loci. Proc. Natl. Acad. Sci. USA 2017, 114, E11257–E11266. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479. [Google Scholar] [CrossRef] [PubMed]
- Dolei, A. The aliens inside us: HERV-W endogenous retroviruses and multiple sclerosis. Mult. Scler. J. 2018, 24, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Dougier-Reynaud, H.L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.-B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human Endogenous Retrovirus Protein Activates Innate Immunity and Promotes Experimental Allergic Encephalomyelitis in Mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef] [PubMed]
- Kessler, A.; Wiesner, M.; Denner, J.; Kämmerer, U.; Vince, G.; Linsenmann, T.; Löhr, M.; Ernestus, R.I.; Hagemann, C. Expression-analysis of the human endogenous retrovirus HERV-K in human astrocytic tumors. BMC Res. Not. 2014, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Radvanyi, L.; Yin, B.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; Li, D.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef] [PubMed]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2011, 69, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.; Heyne, K.; Roemer, K.; Meese, E.; Mayer, J. HERV-K(HML-2) rec and np9 transcripts not restricted to disease but present in many normal human tissues. Mobile DNA 2015, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Dewannieux, M.; Blaise, S.; Heidmann, T. Identification of a Functional Envelope Protein from the HERV-K Family of Human Endogenous Retroviruses. J. Virol. 2005, 79, 15573–15577. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Yasunaga, T.; Miyata, T.; Ushikubo, H. Nucleotide sequence of human endogenous retrovirus genome related to the mouse mammary tumor virus genome. J. Virol. 1986, 60, 589–598. [Google Scholar] [PubMed]
- Giebler, M.; Staege, M.S.; Blauschmidt, S.; Ohm, L.I.; Kraus, M.; Würl, P.; Taubert, H.; Greither, T. Elevated HERV-K Expression in Soft Tissue Sarcoma Is Associated with Worsened Relapse-Free Survival. Front. Microb. 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2013, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Uleri, E.; Ibba, G.; Piu, C.; Caocci, M.; Leoni, S.; Arru, G.; Serra, C.; Sechi, G.; Dolei, A. JC polyomavirus expression and bell-shaped regulation of its SF2/ASF suppressor during the follow-up of multiple sclerosis patients treated with natalizumab. J. Neuro Virol. 2016, 23, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.; Berkhout, B.; Das, A.T. HIV-1 splicing is controlled by local RNA structure and binding of splicing regulatory proteins at the major 5′ splice site. J. Gen. Virol. 2015, 96, 1906–1917. [Google Scholar] [CrossRef] [PubMed]
- Goering, W.; Ribarska, T.; Schulz, W.A. Selective changes of retroelement expression in human prostate cancer. Carcinogenesis 2011, 32, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Hong, Z.; Liu, H.; Chen, X.; Ding, L.; Liu, Z.; Zhou, F.; Yuan, Y. Human Endogenous Retroviruses-K (HML-2) Expression Is Correlated with Prognosis and Progress of Hepatocellular Carcinoma. BioMed Res. Int. 2016, 2016, 8201642. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, U.; Schulz, W.A.; Koch, A.; Niegisch, G.; Goering, W. HERV-K and LINE-1 DNA Methylation and Reexpression in Urothelial Carcinoma. Front. Oncol. 2013, 3, 255. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Plummer, J.B.; Yin, B.; Li, M.; Garza, J.; Radvanyi, L.; Ramondetta, L.M.; Lin, K.; Johanning, G.L.; Tang, D.G.; et al. Cytotoxicity of Human Endogenous Retrovirus K-Specific T Cells toward Autologous Ovarian Cancer Cells. Clin. Can. Res. 2014, 21, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Galindo, R.; Kaplan, M.H.; Contreras-Galindo, A.C.; Gonzalez-Hernandez, M.J.; Ferlenghi, I.; Giusti, F.; Lorenzo, E.; Gitlin, S.D.; Dosik, M.H.; Yamamura, Y.; et al. Characterization of Human Endogenous Retroviral Elements in the Blood of HIV-1-Infected Individuals. J. Virol. 2012, 86, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Maldarelli, F.; Mellors, J.; Coffin, J.M. HIV-1 Infection Leads to Increased Transcription of Human Endogenous Retrovirus HERV-K (HML-2) Proviruses In Vivo but Not to Increased Virion Production. J. Virol. 2014, 88, 11108–11120. [Google Scholar] [CrossRef] [PubMed]
- Zufiría, M.; Gil-Bea, F.J.; Fernández-Torrón, R.; Poza, J.J.; Muñoz-Blanco, J.L.; Rojas-García, R.; Riancho, J.; Munain, A.L.D. ALS: A bucket of genes, environment, metabolism and unknown ingredients. Prog. Neurobiol. 2016, 142, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.W.; Chen, M.T.; Lin, L.T.; Huang, P.I.; Lo, W.L.; Yang, Y.P.; Lu, K.H.; Chen, Y.W.; Chiou, S.H.; Wu, C.W. TDP-43/HDAC6 axis promoted tumor progression and regulated nutrient deprivation-induced autophagy in glioblastoma. Oncotarget 2017, 8, 56612–56625. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Jiao, F.; Song, Z.; Li, S.; Liu, B.; Yang, H.; Zhou, Q.; Li, Z. Regulation of MALAT1 expression by TDP43 controls the migration and invasion of non-small cell lung cancer cells in vitro. Biochem. Biophys. Res. Commun. 2015, 465, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Groote, C.C.D.; Broeckhoven, C.V.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibba, G.; Piu, C.; Uleri, E.; Serra, C.; Dolei, A. Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis. Viruses 2018, 10, 412. https://doi.org/10.3390/v10080412
Ibba G, Piu C, Uleri E, Serra C, Dolei A. Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis. Viruses. 2018; 10(8):412. https://doi.org/10.3390/v10080412
Chicago/Turabian StyleIbba, Gabriele, Claudia Piu, Elena Uleri, Caterina Serra, and Antonina Dolei. 2018. "Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis" Viruses 10, no. 8: 412. https://doi.org/10.3390/v10080412
APA StyleIbba, G., Piu, C., Uleri, E., Serra, C., & Dolei, A. (2018). Disruption by SaCas9 Endonuclease of HERV-Kenv, a Retroviral Gene with Oncogenic and Neuropathogenic Potential, Inhibits Molecules Involved in Cancer and Amyotrophic Lateral Sclerosis. Viruses, 10(8), 412. https://doi.org/10.3390/v10080412