Biochemical and Functional Characterization of Mouse Mammary Tumor Virus Full-Length Pr77Gag Expressed in Prokaryotic and Eukaryotic Cells


 , and
, and 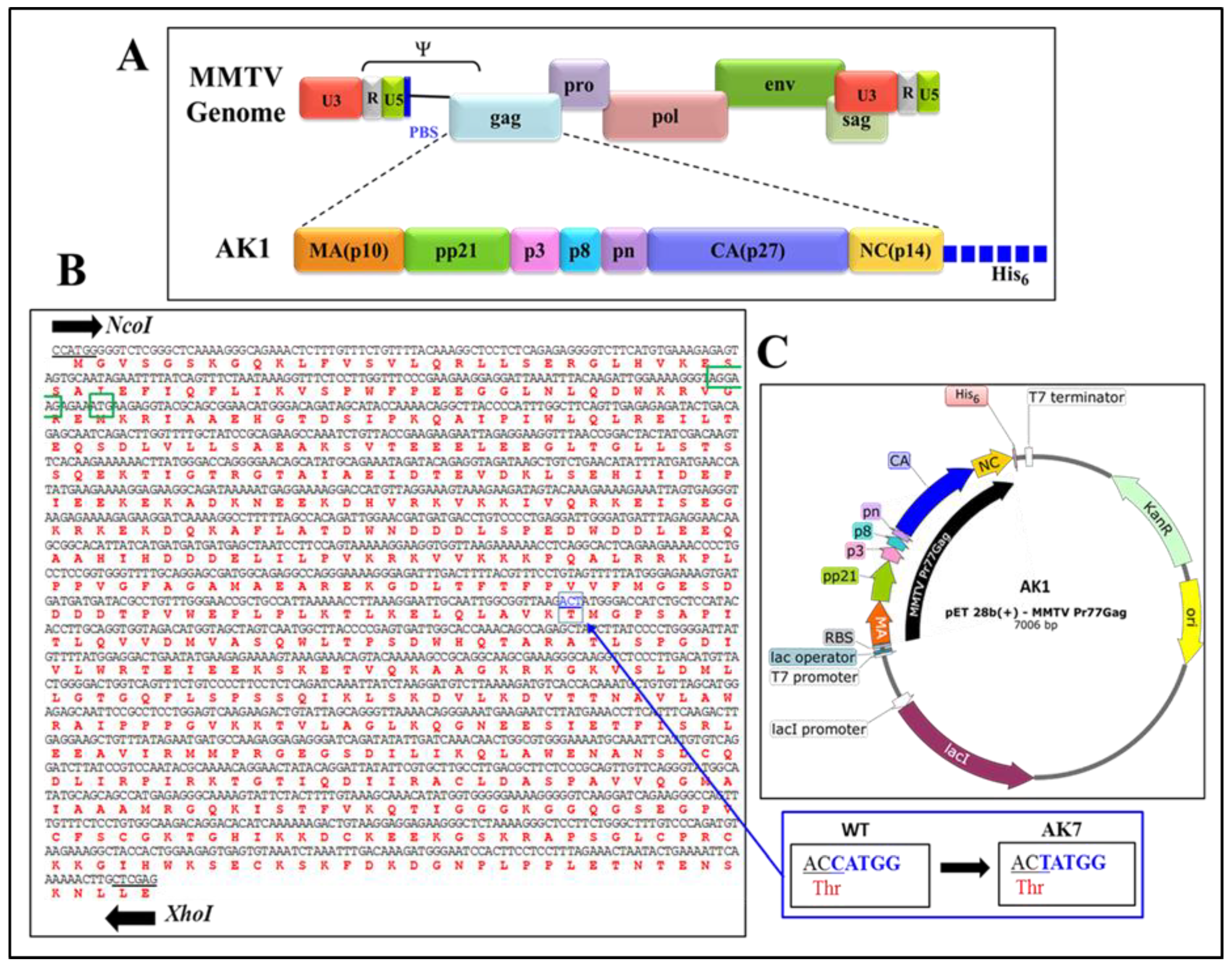{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Nucleotide Numbers
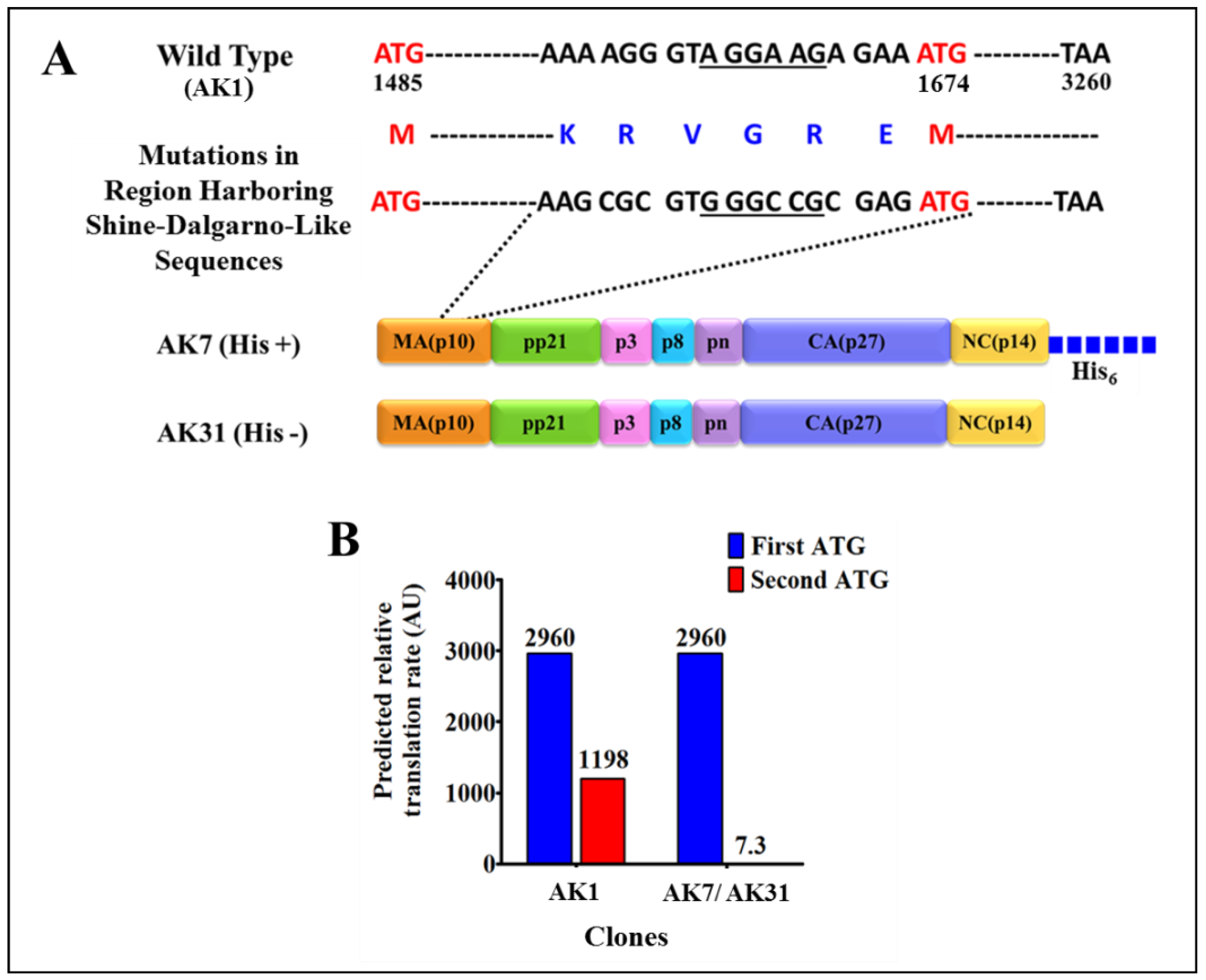
2.2. Full-Length Recombinant Gag Prokaryotic Expression Plasmids
2.3. Full-Length Recombinant Gag Eukaryotic Expression Plasmids
2.4. Escherichia coli Strains and Growth Media
2.5. Expression of Recombinant Full-Length Pr77Gag-His6-Tagged Protein in Bacteria
2.6. Affinity Purification and Gel Filtration Chromatography
2.7. Expression of Recombinant Full-Length Pr77Gag-His6-Tagged Protein in Eukaryotic Cells
2.8. Estimation of RNA Packaging Potential by Reverse Transcriptase PCR
2.9. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis and Western Blotting
2.10. Detection of Prokaryotically-Expressed Virus-Like Particles Using Transmission Electron Microscopy
2.11. In Vitro Assembly of Virus-Like Particles from Bacterially-Expressed Recombinant Full-Length Pr77Gag-His6-Tag Protein
3. Results and Discussion
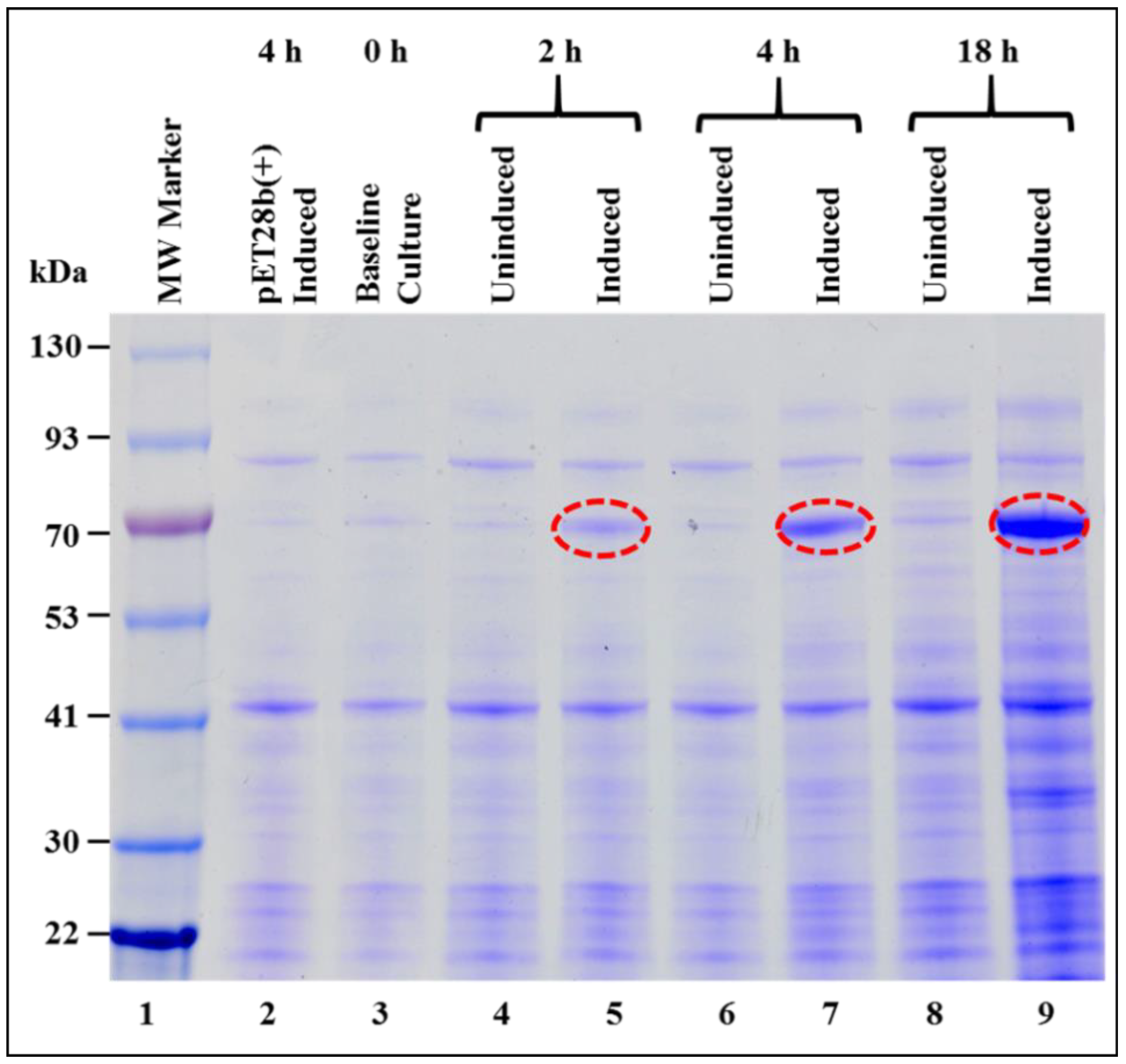
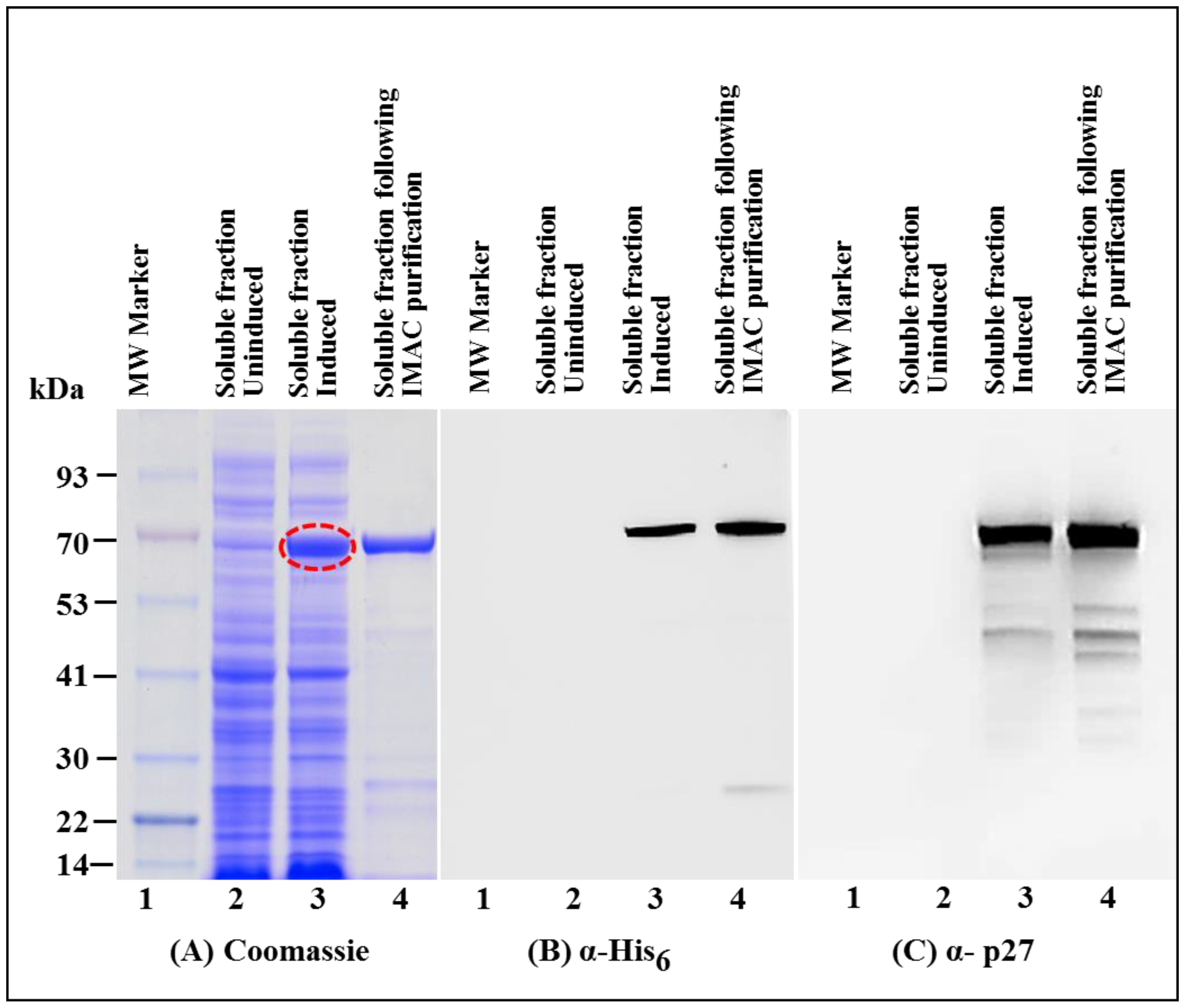
3.1. Successful Expression of Full-Length Recombinant MMTV Pr77Gag-His6-Tagged Protein in Bacteria
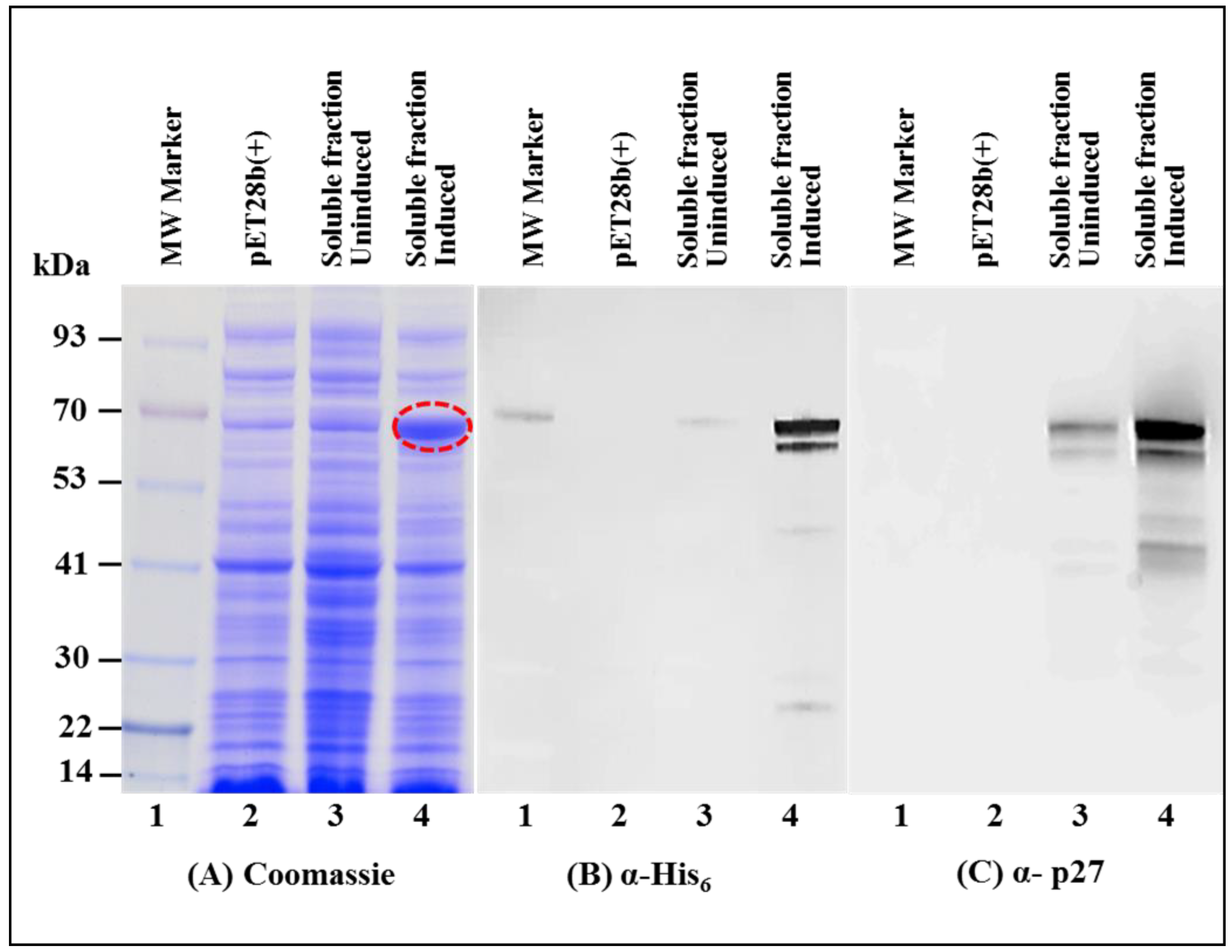
3.2. Full-Length MMTV Pr77Gag-His6-Tagged Fusion Protein Is Expressed in the Soluble Form in Bacteria
3.3. Immobilized Metal Affinity Chromatography Purification of the Soluble Fraction Containing Recombinant Full-Length Pr77Gag-His6-Tagged Fusion Protein
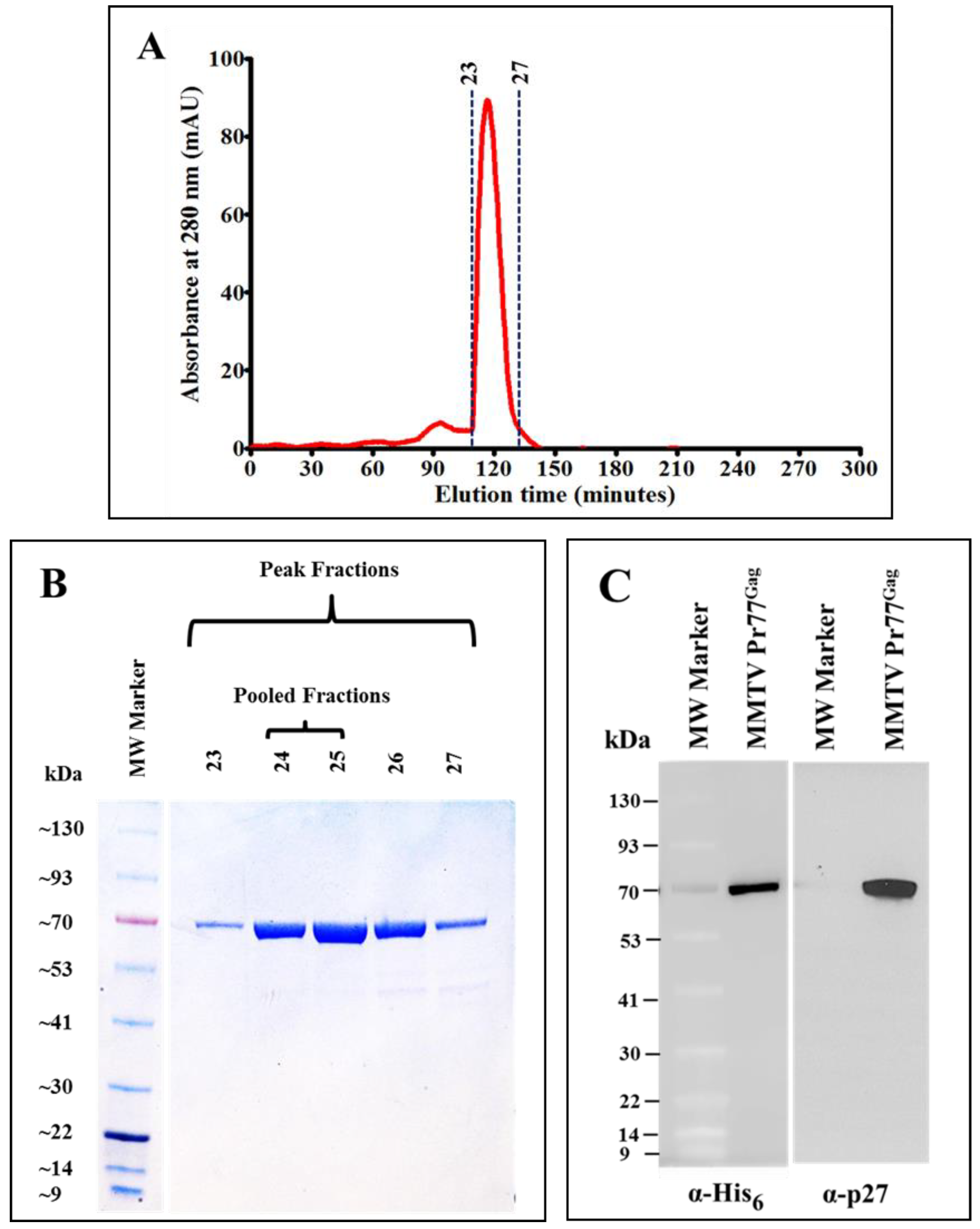
3.4. Gel Filtration Chromatography Purification of the IMAC-Purified Recombinant Full-Length Pr77Gag-His6-Tagged Fusion Protein
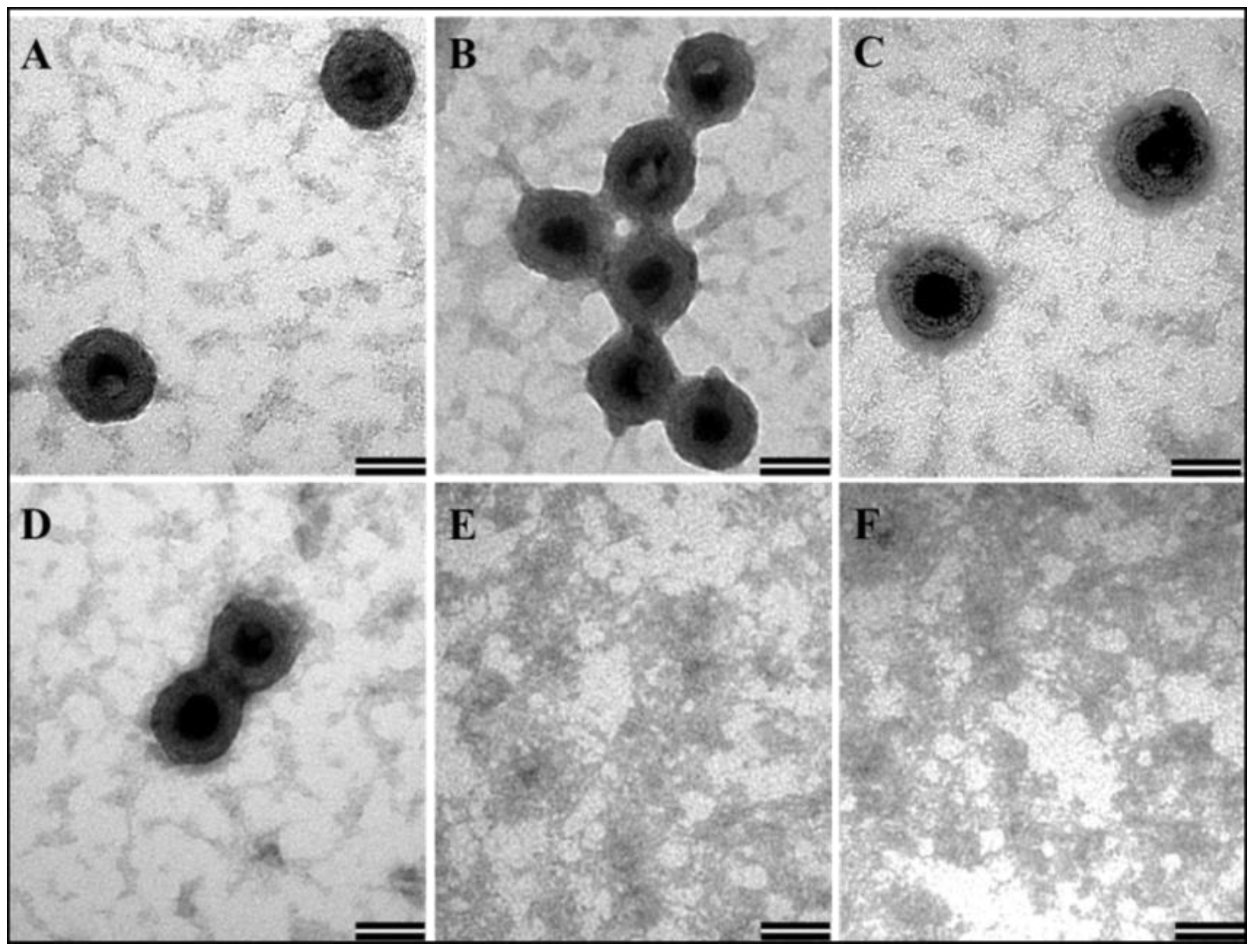
3.5. In Vitro Assembly to Form Virus-Like Particles by the Recombinant Full-Length Pr77Gag-His6-Tagged Fusion Protein
3.6. Recombinant Full-Length MMTV Pr77Gag-His6-Tagged Protein Expressed in Bacteria Can Form Virus-Like Particles
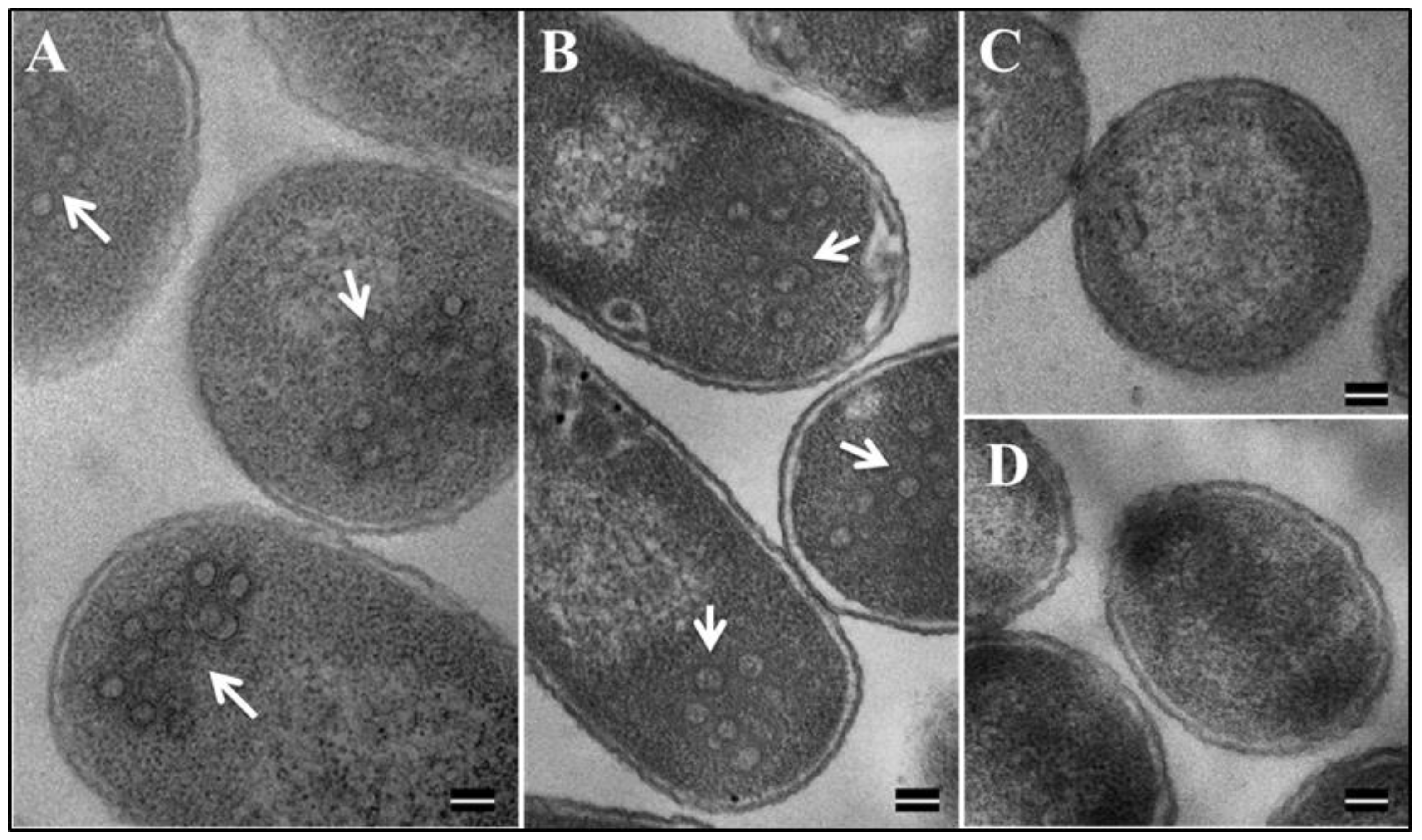
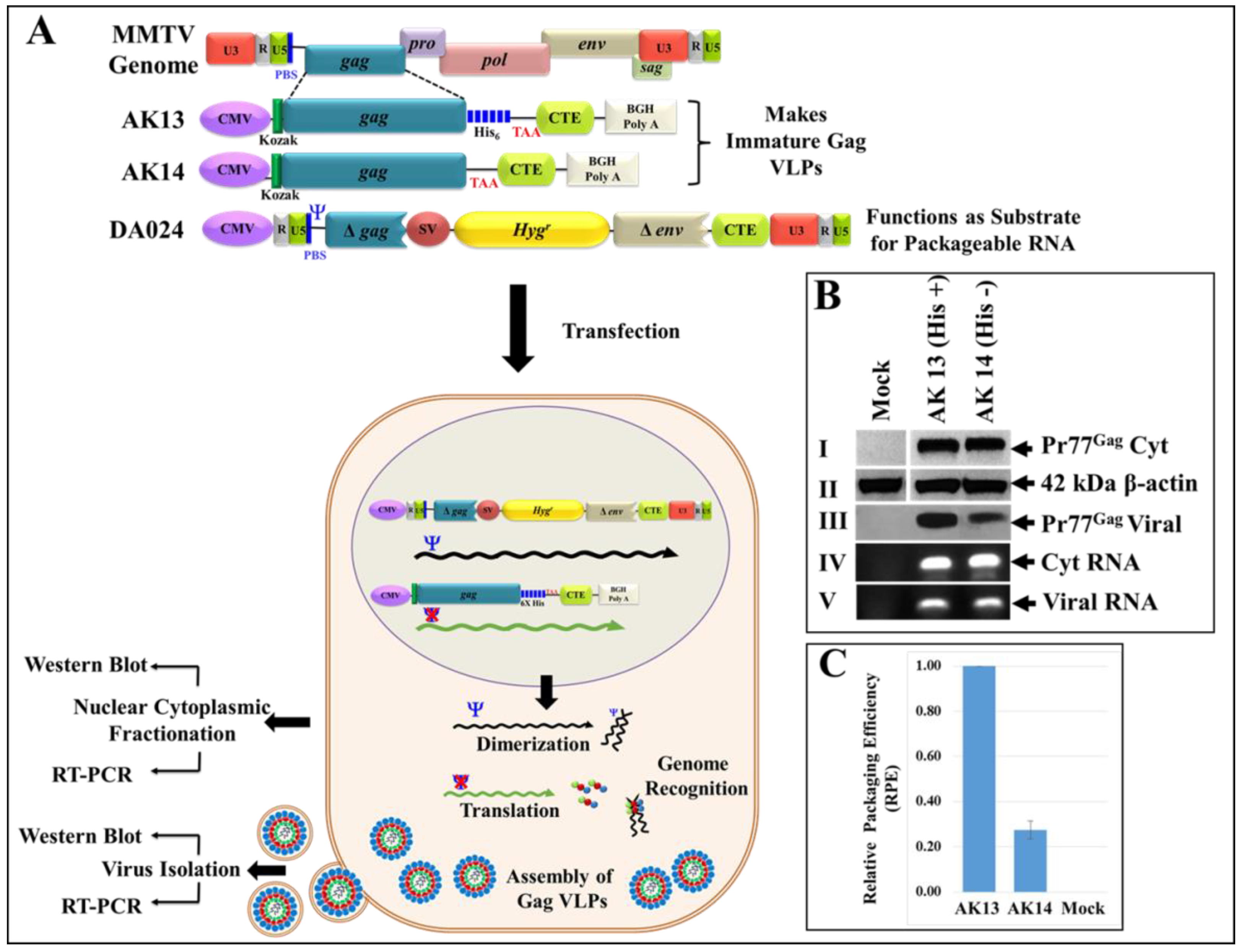
3.7. Eukaryotically-Expressed, Full-Length Recombinant Pr77Gag His6-Tagged Fusion Protein Can Form Virus-Like Particles Competent to Package Unspliced Sub-Genomic RNA
4. Conclusions
Supplementary Materials
Supplementary File 1Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bittner, J.J. Some possible effects of nursing on the mammary gland tumor incidence in mice. Science 1936, 84, 162. [Google Scholar] [CrossRef] [PubMed]
- Cardiff, R.D.; Kenney, N. Mouse mammary tumor biology: A short history. Adv. Cancer Res. 2007, 98, 53–116. [Google Scholar] [CrossRef] [PubMed]
- Duesberg, P.H.; Blair, P.B. Isolation of the nucleic acid of mouse mammary tumor virus (MTV). Proc. Natl. Acad. Sci. USA 1966, 55, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Varmus, H.E.; Bishop, J.M.; Nowinski, R.C.; Sarker, N.H. Mammary Tumour Virus Specific Nucleotide Sequences in Mouse DNA. Nat. New Biol. 1972, 238, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.P.; Golovkina, T.V.; Ross, S.R. Lessons Learned from Mouse Mammary Tumor Virus in Animal Models. ILAR J. 2016, 57, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.E.; Huang, W.; Anwar, T.; Arellano-Garcia, C.; Burman, B.; Guan, J.-L.; Gonzalez, M.E.; Kleer, C.G. MMTV-cre;Ccn6 knockout mice develop tumors recapitulating human metaplastic breast carcinomas. Oncogene 2017, 36, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Konstantoulas, C.J.; Indik, S. Mouse mammary tumor virus-based vector transduces non-dividing cells, enters the nucleus via a TNPO3-independent pathway and integrates in a less biased fashion than other retroviruses. Retrovirology 2014, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Thomson, A.; Needham, M.; Webb, P.; Parker, M. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Res. 1988, 16, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Ruttkowski, B.; Schwab, S.; Peterbauer, T.; Salmons, B.; Günzburg, W.H.; Hohenadl, C. Mouse Mammary Tumor Virus Promoter-Containing Retroviral Promoter Conversion Vectors for Gene-Directed Enzyme Prodrug Therapy are Functional In Vitro and In Vivo. J. Biomed. Biotechnol. 2008, 2008. [Google Scholar] [CrossRef] [PubMed]
- Rouault, F.; Nejad Asl, S.B.; Rungaldier, S.; Fuchs, E.; Salmons, B.; Günzburg, W.H. Promoter complex in the central part of the mouse mammary tumor virus long terminal repeat. J. Virol. 2007, 81, 12572–12581. [Google Scholar] [CrossRef] [PubMed]
- Indik, S. Mouse mammary tumor virus-based vector for efficientand safe transgene delivery into mitotic and non-mitotic cells. Cell Gene Ther. Insights 2016, 2, 589–597. [Google Scholar] [CrossRef]
- Indik, S.; Günzburg, W.H.; Salmons, B.; Rouault, F. Mouse mammary tumor virus infects human cells. Cancer Res. 2005, 65, 6651–6659. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Chadee, A.B.; Byun, H.; Russell, R.; Dudley, J.P. Mapping of the functional boundaries and secondary structure of the mouse mammary tumor virus Rem-responsive element. J. Biol. Chem. 2009, 284, 25642–25652. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Simper, M.S.; Lozano, M.M.; Payne, S.M.; Dudley, J.P. Mouse Mammary Tumor Virus Encodes a Self-Regulatory RNA Export Protein and Is a Complex Retrovirus. J. Virol. 2005, 79, 14737–14747. [Google Scholar] [CrossRef] [PubMed]
- Müllner, M.; Salmons, B.; Günzburg, W.H.; Indik, S. Identification of the Rem-responsive element of mouse mammary tumor virus. Nucleic Acids Res. 2008, 36, 6284–6294. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. Purification, Composition, and Morphology of Virions; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Ali, L.M.; Rizvi, T.A.; Mustafa, F. Cross- and Co-Packaging of Retroviral RNAs and Their Consequences. Viruses 2016, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Comas-Garcia, M.; Davis, S.R.; Rein, A. On the Selective Packaging of Genomic RNA by HIV-1. Viruses 2016, 8, 246. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.; Summers, M.F. How retroviruses select their genomes. Nat. Rev. Microbiol. 2005, 3, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.; Marquet, R.; Paillart, J.-C.; Bernacchi, S. Retroviral RNA Dimerization: From Structure to Functions. Front. Microbiol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.F.; Telesnitsky, A. Retroviral RNA Dimerization and Packaging: The What, How, When, Where, and Why. PLOS Pathog. 2010, 6, e1001007. [Google Scholar] [CrossRef] [PubMed]
- Kaddis Maldonado, R.J.; Parent, L.J. Orchestrating the Selection and Packaging of Genomic RNA by Retroviruses: An Ensemble of Viral and Host Factors. Viruses 2016, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Lever, A.M.L. HIV-1 RNA packaging. Adv. Pharmacol. 2007, 55, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Mailler, E.; Bernacchi, S.; Marquet, R.; Paillart, J.-C.; Vivet-Boudou, V.; Smyth, R.P. The Life-Cycle of the HIV-1 Gag-RNA Complex. Viruses 2016, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Wahab, E.W.; Smyth, R.P.; Mailler, E.; Bernacchi, S.; Vivet-Boudou, V.; Hijnen, M.; Jossinet, F.; Mak, J.; Paillart, J.-C.; Marquet, R. Specific recognition of the HIV-1 genomic RNA by the Gag precursor. Nat. Commun. 2014, 5, 4304. [Google Scholar] [CrossRef] [PubMed]
- Bernacchi, S.; Abd El-Wahab, E.W.; Dubois, N.; Hijnen, M.; Smyth, R.P.; Mak, J.; Marquet, R.; Paillart, J.-C. HIV-1 Pr55Gag binds genomic and spliced RNAs with different affinity and stoichiometry. RNA Biol. 2017, 14, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Smith, M.R.; Jousset, A.-C.; Despons, L.; Laumond, G.; Decoville, T.; Cattenoz, P.; Moog, C.; Jossinet, F.; Mougel, M.; et al. In cell mutational interference mapping experiment (in cell MIME) identifies the 5’ polyadenylation signal as a dual regulator of HIV-1 genomic RNA production and packaging. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Smyth, R.P.; Despons, L.; Huili, G.; Bernacchi, S.; Hijnen, M.; Mak, J.; Jossinet, F.; Weixi, L.; Paillart, J.-C.; von Kleist, M.; et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods 2015, 12, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Salmons, B.; Moritz-Legrand, S.; Garcha, I.; Günzburg, W.H. Construction and characterization of a packaging cell line for MMTV-based conditional retroviral vectors. Biochem. Biophys. Res. Commun. 1989, 159, 1191–1198. [Google Scholar] [CrossRef]
- Rizvi, T.A.; Ali, J.; Phillip, P.S.; Ghazawi, A.; Jayanth, P.; Mustafa, F. Role of a heterologous retroviral transport element in the development of genetic complementation assay for mouse mammary tumor virus (MMTV) replication. Virology 2009, 385, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, F.; Amri, D.A.; Ali, F.A.; Sari, N.A.; Suwaidi, S.A.; Jayanth, P.; Philips, P.S.; Rizvi, T.A. Sequences within Both the 5′ UTR and Gag Are Required for Optimal In Vivo Packaging and Propagation of Mouse Mammary Tumor Virus (MMTV) Genomic RNA. PLoS ONE 2012, 7, e47088. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Aktar, S.J.; Vivet-Boudou, V.; Ali, L.M.; Jabeen, A.; Kalloush, R.M.; Richer, D.; Mustafa, F.; Marquet, R.; Rizvi, T.A. Structural basis of genomic RNA (gRNA) dimerization and packaging determinants of mouse mammary tumor virus (MMTV). Retrovirology 2014, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Jewell, N.A.; Mansky, L.M. In the beginning: Genome recognition, RNA encapsidation and the initiation of complex retrovirus assembly. J. Gen. Virol. 2000, 81, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Aldovini, A.; Young, R.A. Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J. Virol. 1990, 64, 1920–1926. [Google Scholar] [PubMed]
- Dorfman, T.; Luban, J.; Goff, S.P.; Haseltine, W.A.; Göttlinger, H.G. Mapping of functionally important residues of a cysteine-histidine box in the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 1993, 67, 6159–6169. [Google Scholar] [PubMed]
- Gorelick, R.J.; Henderson, L.E.; Hanser, J.P.; Rein, A. Point mutants of Moloney murine leukemia virus that fail to package viral RNA: Evidence for specific RNA recognition by a “zinc finger-like” protein sequence. Proc. Natl. Acad. Sci. USA 1988, 85, 8420–8424. [Google Scholar] [CrossRef] [PubMed]
- Méric, C.; Gouilloud, E.; Spahr, P.F. Mutations in Rous sarcoma virus nucleocapsid protein p12 (NC): Deletions of Cys-His boxes. J. Virol. 1988, 62, 3328–3333. [Google Scholar] [PubMed]
- Poon, D.T.; Wu, J.; Aldovini, A. Charged amino acid residues of human immunodeficiency virus type 1 nucleocapsid p7 protein involved in RNA packaging and infectivity. J. Virol. 1996, 70, 6607–6616. [Google Scholar] [PubMed]
- Lu, K.; Heng, X.; Garyu, L.; Monti, S.; Garcia, E.L.; Kharytonchyk, S.; Dorjsuren, B.; Kulandaivel, G.; Jones, S.; Hiremath, A.; et al. NMR detection of structures in the HIV-1 5’-leader RNA that regulate genome packaging. Science 2011, 334, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Bieniasz, P.D. Analysis of the Initiating Events in HIV-1 Particle Assembly and Genome Packaging. PLOS Pathog. 2010, 6, e1001200. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.F.; Lever, A.M.L. Nonreciprocal Packaging of Human Immunodeficiency Virus Type 1 and Type 2 RNA: A Possible Role for the p2 Domain of Gag in RNA Encapsidation. J. Virol. 1998, 72, 5877–5885. [Google Scholar] [PubMed]
- Roy, B.B.; Russell, R.S.; Turner, D.; Liang, C. The T12I mutation within the SP1 region of Gag restricts packaging of spliced viral RNA into human immunodeficiency virus type 1 with mutated RNA packaging signals and mutated nucleocapsid sequence. Virology 2006, 344, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.S.; Roldan, A.; Detorio, M.; Hu, J.; Wainberg, M.A.; Liang, C. Effects of a Single Amino Acid Substitution within the p2 Region of Human Immunodeficiency Virus Type 1 on Packaging of Spliced Viral RNA. J. Virol. 2003, 77, 12986–12995. [Google Scholar] [CrossRef] [PubMed]
- Tanwar, H.S.; Khoo, K.K.; Garvey, M.; Waddington, L.; Leis, A.; Hijnen, M.; Velkov, T.; Dumsday, G.J.; McKinstry, W.J.; Mak, J. The thermodynamics of Pr55Gag-RNA interaction regulate the assembly of HIV. PLOS Pathog. 2017, 13, e1006221. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.; Summers, M.F. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature 2004, 431, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Garcia, E.L.; King, S.R.; Iyalla, K.; Loeliger, K.; Starck, P.; Syed, S.; Telesnitsky, A.; Summers, M.F. An RNA Structural Switch Regulates Diploid Genome Packaging by Moloney Murine Leukemia Virus. J. Mol. Biol. 2010, 396, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Hizi, A.; Henderson, L.E.; Copeland, T.D.; Sowder, R.C.; Krutzsch, H.C.; Oroszlan, S. Analysis of gag proteins from mouse mammary tumor virus. J. Virol. 1989, 63, 2543–2549. [Google Scholar] [PubMed]
- Hizi, A.; Henderson, L.E.; Copeland, T.D.; Sowder, R.C.; Hixson, C.V.; Oroszlan, S. Characterization of mouse mammary tumor virus gag-pro gene products and the ribosomal frameshift site by protein sequencing. Proc. Natl. Acad. Sci. USA 1987, 84, 7041–7045. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H. Evidence for a precursor-product relationship between intracytoplasmic A particles and mouse mammary tumour virus cores. J. Gen. Virol. 1978, 41, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Tamura, A.; Tsujimura, D. Properties of the intracytoplasmic A particles purified from mouse tumors. Virology 1972, 49, 61–78. [Google Scholar] [CrossRef]
- Barajas, B.C.; Tanaka, M.; Robinson, B.A.; Phuong, D.J.; Chutiraka, K.; Reed, J.C.; Lingappa, J.R. Identifying the assembly intermediate in which Gag first associates with unspliced HIV-1 RNA suggests a novel model for HIV-1 RNA packaging. PLoS Pathog. 2018, 14, e1006977. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.T.; Sherer, N.M. Subcellular Localization of HIV-1 gag-pol mRNAs Regulates Sites of Virion Assembly. J. Virol. 2017, 91, e02315-16. [Google Scholar] [CrossRef] [PubMed]
- Behrens, R.T.; Aligeti, M.; Pocock, G.M.; Higgins, C.A.; Sherer, N.M. Nuclear Export Signal Masking Regulates HIV-1 Rev Trafficking and Viral RNA Nuclear Export. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Blißenbach, M.; Grewe, B.; Konietzny, R.; Grunwald, T.; Überla, K. Rev Proteins of Human and Simian Immunodeficiency Virus Enhance RNA Encapsidation. PLOS Pathog. 2007, 3, e54. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Lainé, S.; Pessel-Vivares, L.; Mougel, M. Cell biology of retroviral RNA packaging. RNA Biol. 2011, 8, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Lingappa, J.R.; Tanaka, M.; Barajas, B.C.; Robinson, B.A.; Phuong, D.J.; Chutiraka, K.; Reed, J.C. HIV-1 initiates genomic RNA packaging in a unique subset of host RNA granules. bioRxiv 2017, 183855. [Google Scholar] [CrossRef]
- Moore, M.D.; Nikolaitchik, O.A.; Chen, J.; Hammarskjöld, M.-L.; Rekosh, D.; Hu, W.-S. Probing the HIV-1 Genomic RNA Trafficking Pathway and Dimerization by Genetic Recombination and Single Virion Analyses. PLOS Pathog. 2009, 5, e1000627. [Google Scholar] [CrossRef] [PubMed]
- Köppe, B.; Menéndez-Arias, L.; Oroszlan, S. Expression and purification of the mouse mammary tumor virus gag-pro transframe protein p30 and characterization of its dUTPase activity. J. Virol. 1994, 68, 2313–2319. [Google Scholar] [PubMed]
- Taube, R.; Loya, S.; Avidan, O.; Perach, M.; Hizi, A. Reverse transcriptase of mouse mammary tumour virus: Expression in bacteria, purification and biochemical characterization. Biochem. J. 1998, 329, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Hook, L.M.; Agafonova, Y.; Ross, S.R.; Turner, S.J.; Golovkina, T.V. Genetics of Mouse Mammary Tumor Virus-Induced Mammary Tumors: Linkage of Tumor Induction to the gag Gene. J. Virol. 2000, 74, 8876–8883. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.; Prasad, S.; Dubay, J.W.; Hunter, E.; Jeang, K.T.; Rekosh, D.; Hammarskjöld, M.L. A small element from the Mason-Pfizer monkey virus genome makes human immunodeficiency virus type 1 expression and replication Rev-independent. Proc. Natl. Acad. Sci. USA 1994, 91, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.C.; Reinhart, L.; Stake, M.S.; Nadaraia-Hoke, S.; Parent, L.J.; Flanagan, J.M. A non-cleavable hexahistidine affinity tag at the carboxyl-terminus of the HIV-1 Pr55Gag polyprotein alters nucleic acid binding properties. Protein Expr. Purif. 2017, 130, 137–145. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, W.J.; Hijnen, M.; Tanwar, H.S.; Sparrow, L.G.; Nagarajan, S.; Pham, S.T.; Mak, J. Expression and purification of soluble recombinant full length HIV-1 Pr55Gag protein in Escherichia coli. Protein Expr. Purif. 2014, 100, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ghazawi, A.; Mustafa, F.; Phillip, P.S.; Jayanth, P.; Ali, J.; Rizvi, T.A. Both the 5′ and 3′ LTRs of FIV contain minor RNA encapsidation determinants compared to the two core packaging determinants within the 5’ untranslated region and gag. Microbes Infect. 2006, 8, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, F.; Ghazawi, A.; Jayanth, P.; Phillip, P.S.; Ali, J.; Rizvi, T.A. Sequences Intervening between the Core Packaging Determinants Are Dispensable for Maintaining the Packaging Potential and Propagation of Feline Immunodeficiency Virus Transfer Vector RNAs. J. Virol. 2005, 79, 13817–13821. [Google Scholar] [CrossRef] [PubMed]
- Purdy, A.; Case, L.; Duvall, M.; Overstrom-Coleman, M.; Monnier, N.; Chervonsky, A.; Golovkina, T. Unique resistance of I/LnJ mice to a retrovirus is due to sustained interferon γ-dependent production of virus-neutralizing antibodies. J. Exp. Med. 2003, 197, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Brookes, S.; Placzek, M.; Moore, R.; Dixon, M.; Dickson, C.; Peters, G. Insertion elements and transitions in cloned mouse mammary tumour virus DNA: Further delineation of the poison sequences. Nucleic Acids Res. 1986, 14, 8231–8245. [Google Scholar] [CrossRef] [PubMed]
- Klikova, M.; Rhee, S.S.; Hunter, E.; Ruml, T. Efficient in vivo and in vitro assembly of retroviral capsids from Gag precursor proteins expressed in bacteria. J. Virol. 1995, 69, 1093–1098. [Google Scholar] [PubMed]
- Shine, J.; Dalgarno, L. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: Complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. USA 1974, 71, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Espah Borujeni, A.; Channarasappa, A.S.; Salis, H.M. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014, 42, 2646–2659. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M. The ribosome binding site calculator. Methods Enzymol. 2011, 498, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.A. E. coli Expression Systems. In Molecular Biology Problem Solver; Gerstein, A.S., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 461–490. ISBN 978-0-471-22390-0. [Google Scholar]
- Affranchino, J.L.; González, S.A. In vitro assembly of the feline immunodeficiency virus Gag polyprotein. Virus Res. 2010, 150, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.; Rein, A. In Vitro Assembly Properties of Human Immunodeficiency Virus Type 1 Gag Protein Lacking the p6 Domain. J. Virol. 1999, 73, 2270–2279. [Google Scholar] [PubMed]
- Campbell, S.; Vogt, V.M. In vitro assembly of virus-like particles with Rous sarcoma virus Gag deletion mutants: Identification of the p10 domain as a morphological determinant in the formation of spherical particles. J. Virol. 1997, 71, 4425–4435. [Google Scholar] [PubMed]
- Ehrlich, L.S.; Agresta, B.E.; Carter, C.A. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 1992, 66, 4874–4883. [Google Scholar] [PubMed]
- Sakalian, M.; Parker, S.D.; Weldon, R.A.; Hunter, E. Synthesis and assembly of retrovirus Gag precursors into immature capsids in vitro. J. Virol. 1996, 70, 3706–3715. [Google Scholar] [PubMed]
- Sakalian, M.; Hunter, E. Separate Assembly and Transport Domains within the Gag Precursor of Mason-Pfizer Monkey Virus. J. Virol. 1999, 73, 8073–8082. [Google Scholar] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chameettachal, A.; Pillai, V.N.; Ali, L.M.; Pitchai, F.N.N.; Ardah, M.T.; Mustafa, F.; Marquet, R.; Rizvi, T.A. Biochemical and Functional Characterization of Mouse Mammary Tumor Virus Full-Length Pr77Gag Expressed in Prokaryotic and Eukaryotic Cells. Viruses 2018, 10, 334. https://doi.org/10.3390/v10060334
Chameettachal A, Pillai VN, Ali LM, Pitchai FNN, Ardah MT, Mustafa F, Marquet R, Rizvi TA. Biochemical and Functional Characterization of Mouse Mammary Tumor Virus Full-Length Pr77Gag Expressed in Prokaryotic and Eukaryotic Cells. Viruses. 2018; 10(6):334. https://doi.org/10.3390/v10060334
Chicago/Turabian StyleChameettachal, Akhil, Vineeta Narayana Pillai, Lizna Mohamed Ali, Fathima Nuzra Nagoor Pitchai, Mustafa Taleb Ardah, Farah Mustafa, Roland Marquet, and Tahir Aziz Rizvi. 2018. "Biochemical and Functional Characterization of Mouse Mammary Tumor Virus Full-Length Pr77Gag Expressed in Prokaryotic and Eukaryotic Cells" Viruses 10, no. 6: 334. https://doi.org/10.3390/v10060334
APA StyleChameettachal, A., Pillai, V. N., Ali, L. M., Pitchai, F. N. N., Ardah, M. T., Mustafa, F., Marquet, R., & Rizvi, T. A. (2018). Biochemical and Functional Characterization of Mouse Mammary Tumor Virus Full-Length Pr77Gag Expressed in Prokaryotic and Eukaryotic Cells. Viruses, 10(6), 334. https://doi.org/10.3390/v10060334