K15 Protein of Kaposi’s Sarcoma Herpesviruses Increases Endothelial Cell Proliferation and Migration through Store-Operated Calcium Entry


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Transfections
2.3. Production and Purification of Lentiviral Virions
2.4. Measurement of Cytosolic Ca2+
2.5. Western Blotting
2.6. Cell-Counting Kit-8 (CCK-8) Assay and Wound Scratch Assays
2.7. Statistical Analysis
3. Results
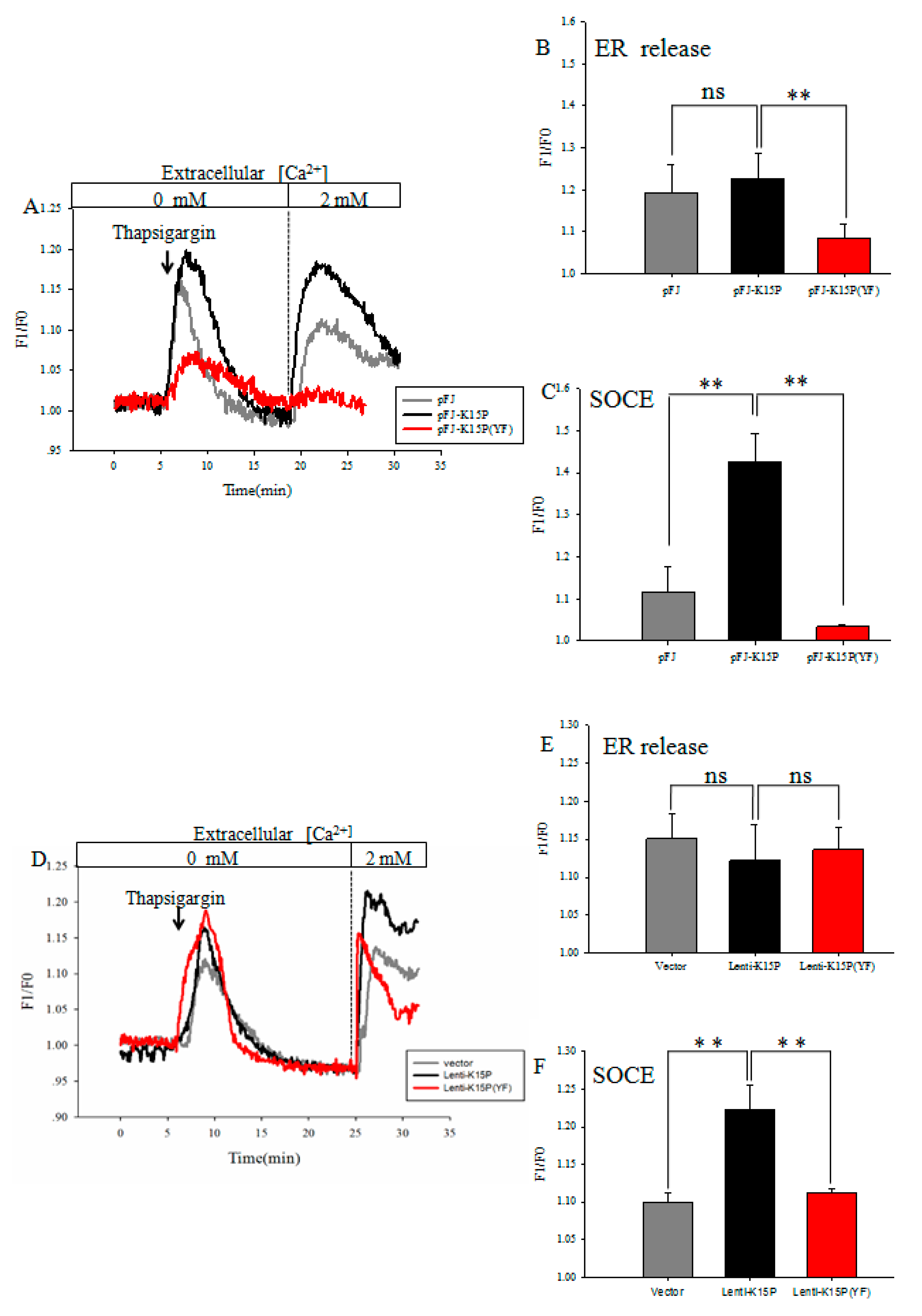
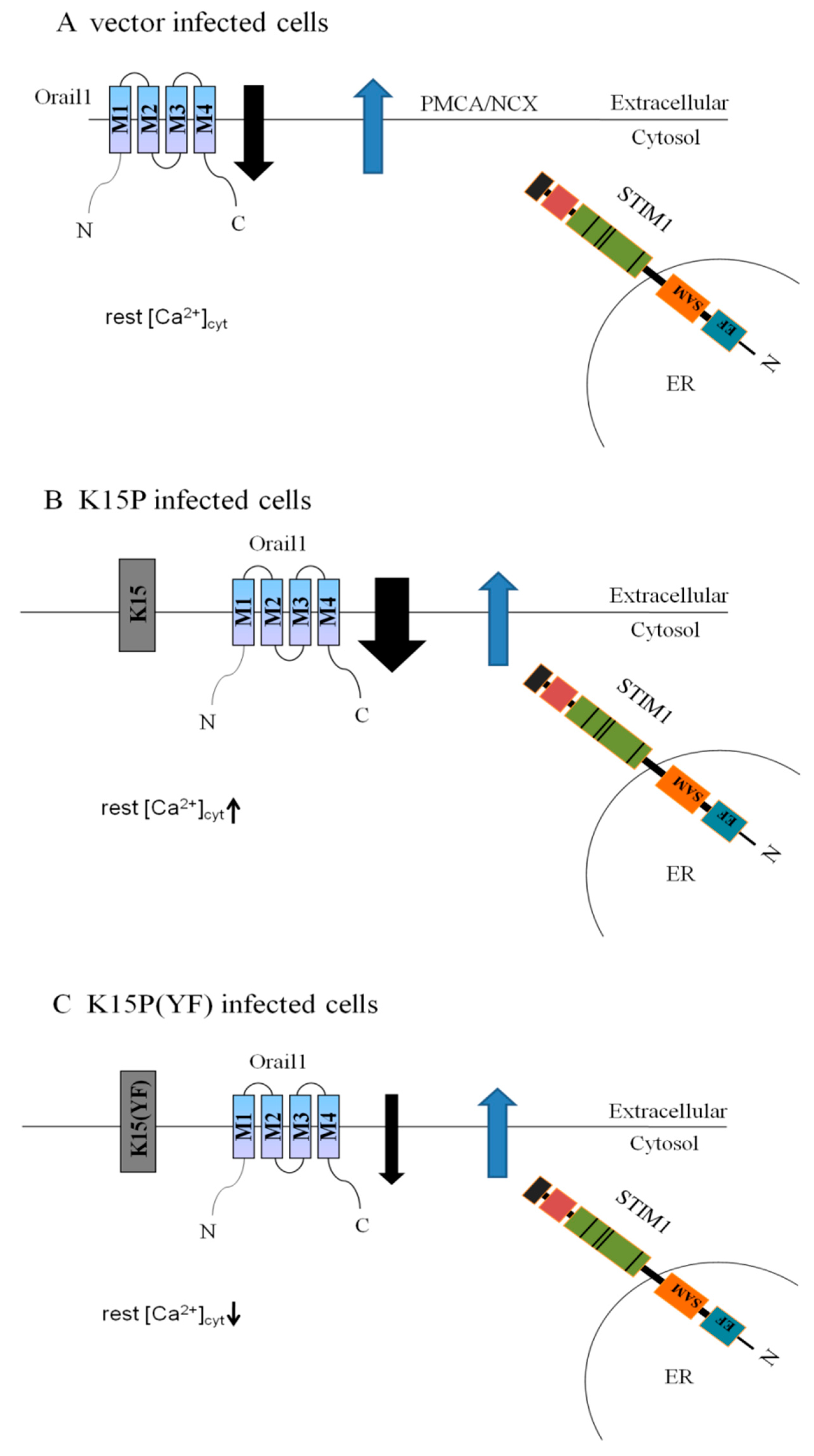
3.1. K15P Amplified Thapsigargin-Stimulated SOCE in HEK-293T and EA.hy926 Cells
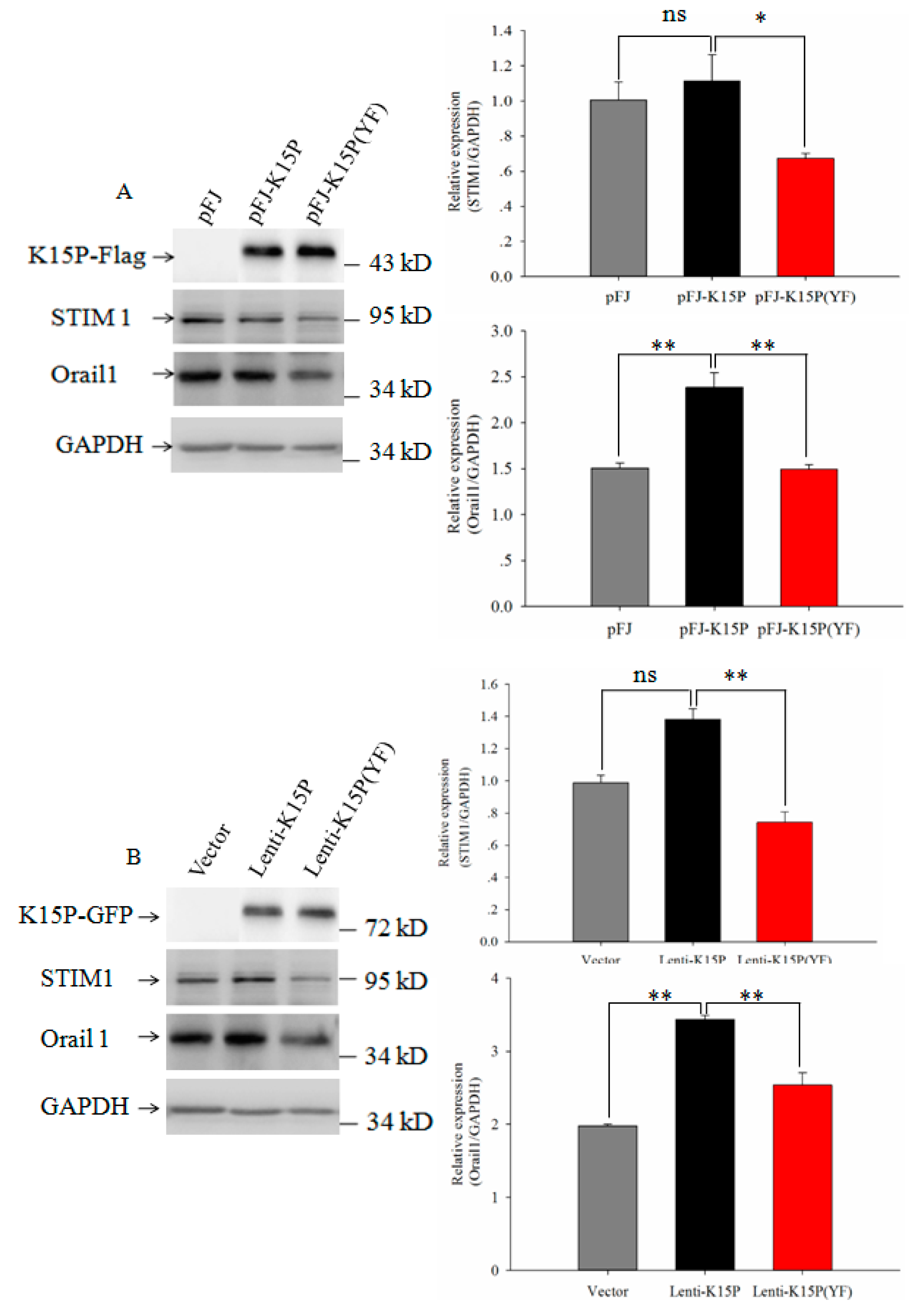
3.2. Expression of STIM1 and Orail1in HEK-293T and EA.hy926 Cells
3.3. K15P Increased Cell Proliferation and Migration
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Cui, H.Y.; Liu, Y.M.; Zhao, P.; Zhang, Y.; Fu, Z.G.; Chen, Z.N.; Jiang, J.L. CD147 promotes Src-dependent activation of Rac1 signaling through STAT3/Dock8 during the motility of hepatocellular carcinoma cells. Oncotarget 2015, 6, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Kohn, E.C. Invasion and metastasis: Biology and clinical potential. Pharmacol. Ther. 1991, 52, 235–244. [Google Scholar] [CrossRef]
- Goncalves, P.H.; Uldrick, T.S.; Yarchoan, R. HIV-associated Kaposi Sarcoma and related diseases. AIDS 2017, 31, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Jemal, A. Cancer in Africa 2012. Cancer Epidemiol. Biomark. Prev. 2014, 23, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in aids-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Moore, P. Twenty years of KSHV. Viruses 2014, 6, 4258–4264. [Google Scholar] [CrossRef] [PubMed]
- Gramolelli, S.; Schulz, T.F. The role of Kaposi sarcoma-associated herpesvirus in the pathogenesis of Kaposi sarcoma. J. Pathol. 2015, 235, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Wang, L.; Pietrek, M.; Brinkmann, M.M.; Havemeier, A.; Fischer, I.; Hillenbrand, B.; Dittrich-Breiholz, O.; Kracht, M.; Chanas, S.; Blackbourn, D.J.; et al. Identification and functional characterization of a spliced rhesus rhadinovirus gene with homology to the K15 gene of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2009, 90, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Pietrek, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Modulation of host gene expression by the K15 protein of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.H.; Choi, Y.K.; Choi, J.K. Multi-transmembrane protein k15 of Kaposi’s sarcoma-associated herpesvirus targets Lyn kinase in the membrane raft and induces NFAT/AP1 activities. Exp. Mol. Med. 2008, 40, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.D.; Brinkmann, M.M.; Pietrek, M.; Ottinger, M.; Dittrich-Breiholz, I.O.; Kracht, M.; Schulz, T.F. Functional characterization of the M-type K15-encoded membrane protein of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2007, 88, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Schulz, T.F. Kshv non-structural membrane proteins involved in the activation of intracellular signaling pathways and the pathogenesis of Kaposi’s sarcoma. Curr. Opin. Virol. 2016, 20, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Havemeier, A.; Gramolelli, S.; Pietrek, M.; Jochmann, R.; Sturzl, M.; Schulz, T.F. Activation of NF-κB by the Kaposi’s sarcoma-associated herpesvirus K15 protein involves recruitment of the NF-κB-inducing kinase, IκB kinases, and phosphorylation of p65. J. Virol. 2014, 88, 13161–13172. [Google Scholar] [CrossRef] [PubMed]
- Pietrek, M.; Brinkmann, M.M.; Glowacka, I.; Enlund, A.; Havemeier, A.; Dittrich-Breiholz, O.; Kracht, M.; Lewitzky, M.; Saksela, K.; Feller, S.M.; et al. Role of the Kaposi’s sarcoma-associated herpesvirus K15 SH3 binding site in inflammatory signaling and B-cell activation. J. Virol. 2010, 84, 8231–8240. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Glenn, M.; Rainbow, L.; Kieser, A.; Henke-Gendo, C.; Schulz, T.F. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 2003, 77, 9346–9358. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015, 7, 72–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Yuan, H.; Jeon, H.; Zhu, Y.; Yoo, S.; Shi, S.; Krueger, B.; Renne, R.; Lu, C.; Jung, J.U.; et al. Human mesenchymal stem cells of diverse origins support persistent infection with Kaposi’s sarcoma-associated herpesvirus and manifest distinct angiogenic, invasive, and transforming phenotypes. mBio 2016, 7, e02109-15. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Minamitani, T.; Ma, Y.; Zhou, H.; Kida, H.; Tsai, C.Y.; Obana, M.; Okuzaki, D.; Fujio, Y.; Kumanogoh, A.; Zhao, B.; et al. Mouse model of Epstein-barr virus LMP1- and LMP2A-driven germinal center B-cell lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4751–4756. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Subramanian, A.; Sheen, T.S.; Tsai, C.H.; Golub, T.R.; Thorley-Lawson, D.A. Epstein-barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: Possible role in nasopharyngeal carcinoma metastasis. J. Virol. 2005, 79, 15430–15442. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, T.; Weber, T.; Kracker, S.; Sommermann, T.; Rajewsky, K.; Yasuda, T. Mouse model for acute Epstein-barr virus infection. Proc. Natl. Acad. Sci. USA 2016, 113, 13821–13826. [Google Scholar] [CrossRef] [PubMed]
- Steinbruck, L.; Gustems, M.; Medele, S.; Schulz, T.F.; Lutter, D.; Hammerschmidt, W. K1 and K15 of Kaposi’s sarcoma-associated herpesvirus are partial functional homologues of latent membrane protein 2A of Epstein-barr virus. J. Virol. 2015, 89, 7248–7261. [Google Scholar] [CrossRef] [PubMed]
- Glenn, M.; Rainbow, L.; Aurade, F.; Davison, A.; Schulz, T.F. Identification of a spliced gene from Kaposi’s sarcoma-associated herpesvirus encoding a protein with similarities to latent membrane proteins 1 and 2A of Epstein-barr virus. J. Virol. 1999, 73, 6953–6963. [Google Scholar] [PubMed]
- Tsai, Y.H.; Wu, M.F.; Wu, Y.H.; Chang, S.J.; Lin, S.F.; Sharp, T.V.; Wang, H.W. The M type K15 protein of Kaposi’s sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J. Virol. 2009, 83, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. The molecular choreography of a store-operated calcium channel. Nature 2007, 446, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Sztretye, M.; Geyer, N.; Vincze, J.; Al-Gaadi, D.; Olah, T.; Szentesi, P.; Kis, G.; Antal, M.; Balatoni, I.; Csernoch, L.; et al. Soce is important for maintaining sarcoplasmic calcium content and release in skeletal muscle fibers. Biophys. J. 2017, 113, 2496–2507. [Google Scholar] [CrossRef] [PubMed]
- Dellis, O.; Arbabian, A.; Papp, B.; Rowe, M.; Joab, I.; Chomienne, C. Epstein-barr virus latent membrane protein 1 increases calcium influx through store-operated channels in b lymphoid cells. J. Biol. Chem. 2011, 286, 18583–18592. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, J.; Si, Y.; Kanada, M.; Zhang, Z.; Terakawa, S.; Watanabe, H. Blockage of LMP1-modulated store-operated Ca(2+) entry reduces metastatic potential in nasopharyngeal carcinoma cell. Cancer Lett. 2015, 360, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhu, J.; Zhang, J.; Jiang, F.; Wang, Z.; Zhang, Y.; Li, J.; Huang, D.; Ke, D.; Ma, R.; et al. Attenuated mesangial cell proliferation related to store-operated Ca2+ entry in aged rat: The role of STIM 1 and ORAI 1. Age 2013, 35, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Yoo, S.M.; Choi, H.S.; Mun, J.Y.; Kang, H.G.; Lee, J.; Park, J.; Gao, S.J.; Lee, M.S. Extracellular vesicles from KSHV-infected endothelial cells activate the complement system. Oncotarget 2017, 8, 99841–99860. [Google Scholar] [CrossRef] [PubMed]
- Mularoni, A.; Gallo, A.; Riva, G.; Barozzi, P.; Miele, M.; Cardinale, G.; Vizzini, G.; Volpes, R.; Grossi, P.; Di Carlo, D.; et al. Successful treatment of kaposi sarcoma-associated herpesvirus inflammatory cytokine syndrome after kidney-liver transplant: Correlations with the human herpesvirus 8 mirnome and specific T cell response. Am. J. Transplant. 2017, 17, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Regamey, N.; Tamm, M.; Wernli, M.; Witschi, A.; Thiel, G.; Cathomas, G.; Erb, P. Transmission of human herpesvirus 8 infection from renal-transplant donors to recipients. N. Engl. J. Med. 1998, 339, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Vart, R.J.; Nikitenko, L.L.; Lagos, D.; Trotter, M.W.B.; Cannon, M.; Bourboulia, D.; Gratrix, F.; Takeuchi, Y.; Boshoff, C. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoletin-2 expression in lymphatic endothelial cells. Cancer Res. 2007, 67, 4042–4051. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Nithianantham, S.; Chandran, B.; Yu, X.L.; Weinberg, A.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induces rapid release of angiopoietin-2 from endothelial cells. J. Virol. 2013, 87, 6326–6335. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Yan, F.; Ben Major, M.; Damania, B. Modulation of kaposi’s sarcoma-associated herpesvirus interleukin-6 function by hypoxia-upregulated protein 1. J. Virol. 2014, 88, 9429–9441. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Berna-Erro, A.; Woodard, G.E.; Rosado, J.A. ORAIS and STIMS: Physiological mechanisms and disease. J. Cell. Mol. Med. 2012, 16, 407–424. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Xu, C.; Wang, L.; Shen, B.; Wang, L. K15 Protein of Kaposi’s Sarcoma Herpesviruses Increases Endothelial Cell Proliferation and Migration through Store-Operated Calcium Entry. Viruses 2018, 10, 282. https://doi.org/10.3390/v10060282
Chen W, Xu C, Wang L, Shen B, Wang L. K15 Protein of Kaposi’s Sarcoma Herpesviruses Increases Endothelial Cell Proliferation and Migration through Store-Operated Calcium Entry. Viruses. 2018; 10(6):282. https://doi.org/10.3390/v10060282
Chicago/Turabian StyleChen, Wei, Changqing Xu, Liuqing Wang, Bing Shen, and Linding Wang. 2018. "K15 Protein of Kaposi’s Sarcoma Herpesviruses Increases Endothelial Cell Proliferation and Migration through Store-Operated Calcium Entry" Viruses 10, no. 6: 282. https://doi.org/10.3390/v10060282
APA StyleChen, W., Xu, C., Wang, L., Shen, B., & Wang, L. (2018). K15 Protein of Kaposi’s Sarcoma Herpesviruses Increases Endothelial Cell Proliferation and Migration through Store-Operated Calcium Entry. Viruses, 10(6), 282. https://doi.org/10.3390/v10060282