Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Antibodies and Chemicals
2.3. Plasmid Construction
2.4. Viral Infection
2.5. Lentiviral Particle Production and Target Cell Transduction
2.6. Plaque Assay
2.7. Confocal Microscopy
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
3.1. ZIKV Infection Induces Autophagy in HUVEC
3.2. Increased Levels of Autophagic Flux in ZIKV-Infected HUVEC
3.3. Pharmacological Inhibition of Autophagy Reduces ZIKV Production in HUVEC
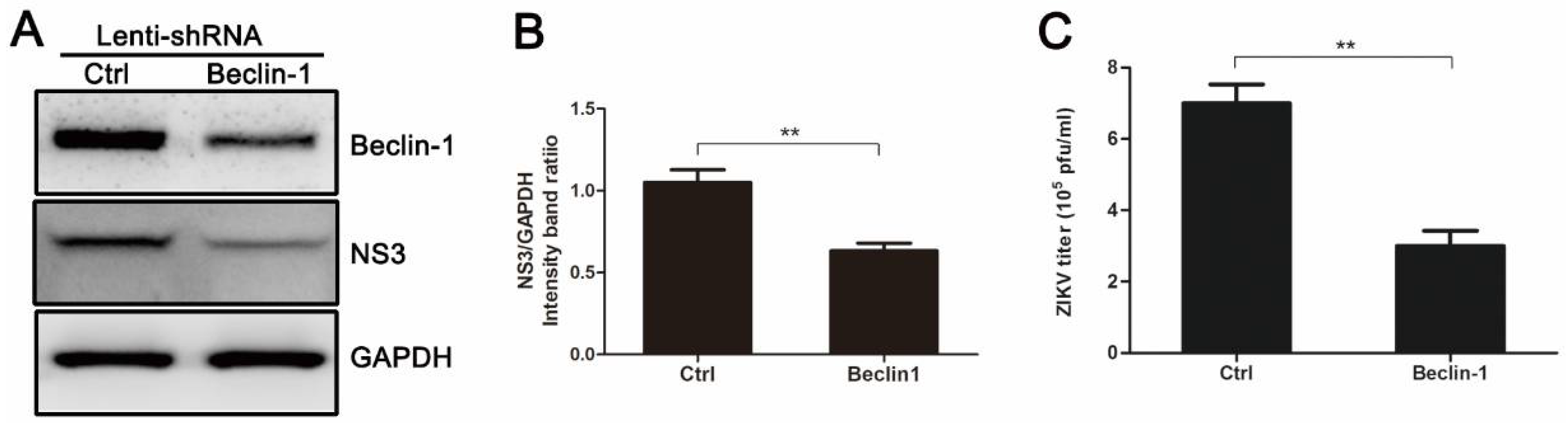
3.4. Knockdown of Autophagy-Related Genes (ATGs) Reduces ZIKV Production in HUVEC
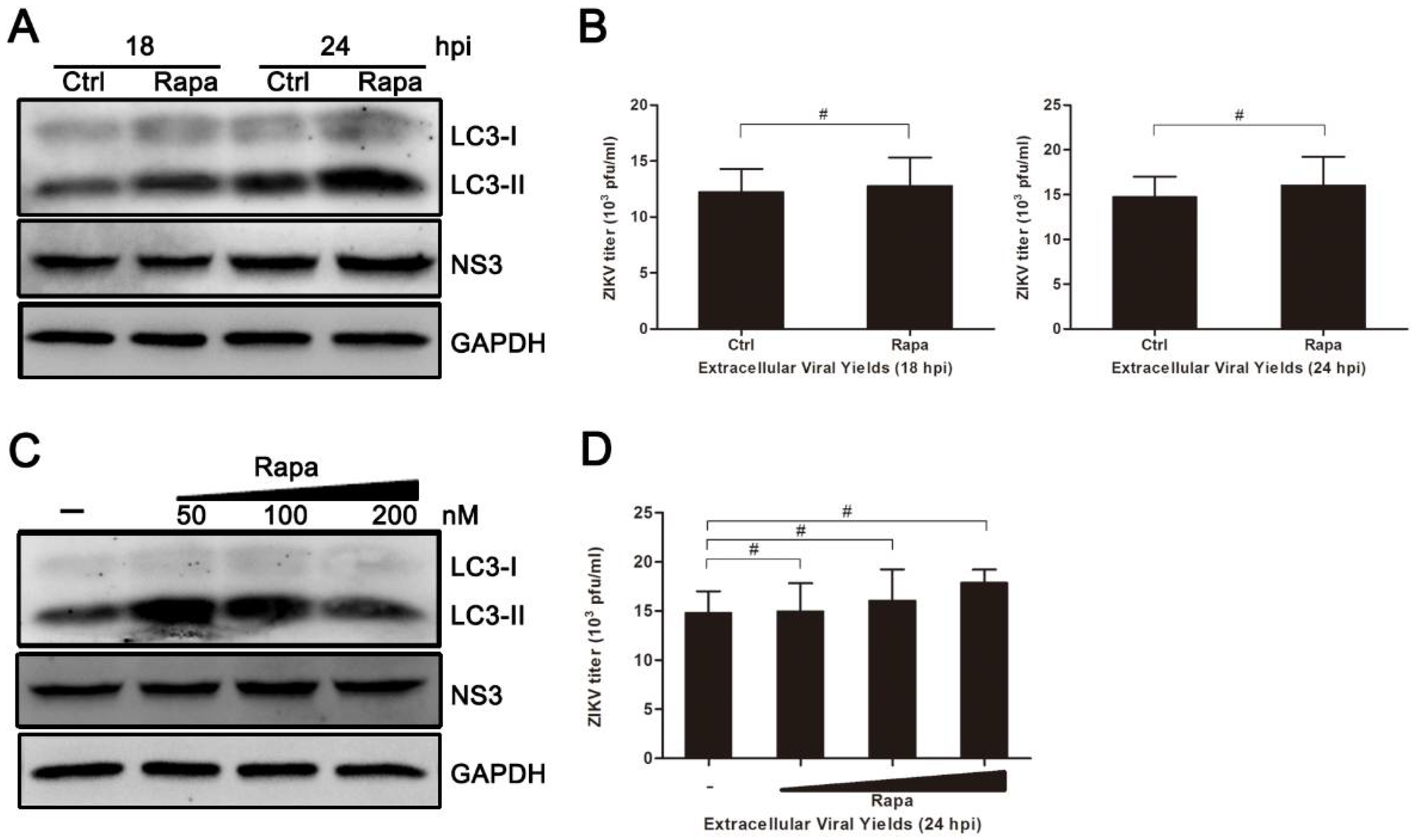
3.5. Rapamycin Does Not Promote ZIKV Production in HUVEC
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.A. Zika virus (II). Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521. [Google Scholar] [CrossRef]
- Musso, D.; Nilles, E.J.; Cao-Lormeau, V.M. Rapid spread of emerging Zika virus in the Pacific area. Clin. Microbiol. Infect. Dis. 2014, 20, O595–O596. [Google Scholar] [CrossRef] [PubMed]
- Chouin-Carneiro, T.; Vega-Rua, A.; Vazeille, M.; Yebakima, A.; Girod, R.; Goindin, D.; Dupont-Rouzeyrol, M.; Lourenco-de-Oliveira, R.; Failloux, A.B. Differential Susceptibilities of Aedes aegypti and Aedes albopictus from the Americas to Zika Virus. PLoS Negl. Trop. Dis. 2016, 10, e0004543. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.; de Sequeira, P.C.; de Mendonca, M.C.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Musso, D.; Roche, C.; Robin, E.; Nhan, T.; Teissier, A.; Cao-Lormeau, V.M. Potential sexual transmission of Zika virus. Emerg. Infect. Dis. 2015, 21, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika Virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Broutet, N.; Krauer, F.; Riesen, M.; Khalakdina, A.; Almiron, M.; Aldighieri, S.; Espinal, M.; Low, N.; Dye, C. Zika Virus as a Cause of Neurologic Disorders. N. Engl. J. Med. 2016, 374, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.P.; Guillaumot, L.; Yug, L.; Saweyog, S.C.; Tided, M.; Machieng, P.; Pretrick, M.; Marfel, M.; Griggs, A.; Bel, M.; et al. Aedes hensilli as a Potential Vector of Chikungunya and Zika Viruses. PLoS Negl. Trop. Dis. 2014, 8, e3188. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.J.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.T.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Bai, B.; Yu, B.; Wang, T.; Wang, H.; Wang, Y.; Li, H.; Tong, L.; Wang, Y.; Zhang, F.; et al. Coxsackievirus B3 induces autophagic response in cardiac myocytes in vivo. Biochemistry 2015, 80, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Chava, S.; Aydin, Y.; Chandra, P.K.; Ferraris, P.; Chen, W.; Balart, L.A.; Wu, T.; Garry, R.F. Hepatitis C Virus Infection Induces Autophagy as a Prosurvival Mechanism to Alleviate Hepatic ER-Stress Response. Viruses 2016, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced autophagy protects cells against death and enhances virus replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Li, Z.L.; Yuan, S. The Role of Secretory Autophagy in Zika Virus Transfer through the Placental Barrier. Front. Cell. Infect. Microbiol. 2016, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.T.; Qin, Z.L.; Ren, H.; Zhao, P.; Qi, Z.T. Inhibition of IRS-1 by hepatitis C virus infection leads to insulin resistance in a PTEN-dependent manner. Virol. J. 2015, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Delbruck, M. The Growth of Bacteriophage and Lysis of the Host. J. Gen. Physiol. 1940, 23, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhou, Z.; Jiang, K.; Yu, S.; Jia, L.; Wu, Y.; Liu, Y.; Meng, S.; Ding, C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch. Virol. 2012, 157, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.L.; Zhao, P.; Cao, M.M.; Qi, Z.T. siRNAs targeting terminal sequences of the SARS-associated coronavirus membrane gene inhibit M protein expression through degradation of M mRNA. J. Virol. Methods 2007, 145, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.L.; Zhao, P.; Zhang, X.L.; Yu, J.G.; Cao, M.M.; Zhao, L.J.; Luan, J.; Qi, Z.T. Silencing of SARS-CoV spike gene by small interfering RNA in HEK 293T cells. Biochem. Biophys. Res. Commun. 2004, 324, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Yamori, T. Phosphatidylinositol 3-kinase inhibitors: Promising drug candidates for cancer therapy. Cancer Sci. 2008, 99, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G.; et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. Human Cytomegalovirus Infection Maintains mTOR Activity and Its Perinuclear Localization during Amino Acid Deprivation. J. Virol. 2011, 85, 9369–9376. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The TORrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Shrivastava, S.; Meyer, K.; Ray, R.B.; Ray, R. Hepatitis C Virus Activates the mTOR/S6K1 Signaling Pathway in Inhibiting IRS-1 Function for Insulin Resistance. J. Virol. 2012, 86, 6315–6322. [Google Scholar] [CrossRef] [PubMed]
- Joubert, P.E.; Werneke, S.W.; de la Calle, C.; Guivel-Benhassine, F.; Giodini, A.; Peduto, L.; Levine, B.; Schwartz, O.; Lenschow, D.J.; Albert, M.L. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 2012, 209, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.; Flint, M.; Nichol, S.T.; Spiropoulou, C.F. Host mTORC1 Signaling Regulates Andes Virus Replication. J. Virol. 2013, 87, 912. [Google Scholar] [CrossRef] [PubMed]
- Stohr, S.; Costa, R.; Sandmann, L.; Westhaus, S.; Pfaender, S.; Anggakusuma; Dazert, E.; Meuleman, P.; Vondran, F.W.R.; Manns, M.P.; et al. Host cell mTORC1 is required for HCV RNA replication. Gut 2016, 65, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10, 259. https://doi.org/10.3390/v10050259
Peng H, Liu B, Yves TD, He Y, Wang S, Tang H, Ren H, Zhao P, Qi Z, Qin Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses. 2018; 10(5):259. https://doi.org/10.3390/v10050259
Chicago/Turabian StylePeng, Haoran, Bin Liu, Toure Doueu Yves, Yanhua He, Shijie Wang, Hailin Tang, Hao Ren, Ping Zhao, Zhongtian Qi, and Zhaoling Qin. 2018. "Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells" Viruses 10, no. 5: 259. https://doi.org/10.3390/v10050259
APA StylePeng, H., Liu, B., Yves, T. D., He, Y., Wang, S., Tang, H., Ren, H., Zhao, P., Qi, Z., & Qin, Z. (2018). Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses, 10(5), 259. https://doi.org/10.3390/v10050259