Human Parainfluenza Virus Type 3 Matrix Protein Reduces Viral RNA Synthesis of HPIV3 by Regulating Inclusion Body Formation

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Plasmid Constructs
2.3. In Vitro Minigenome Assay of HPIV3
2.4. RNA Extraction and RT-PCR
2.5. Immunofluorescence Assay
2.6. Transfection and Recovery of Recombinant HPIV3
3. Results
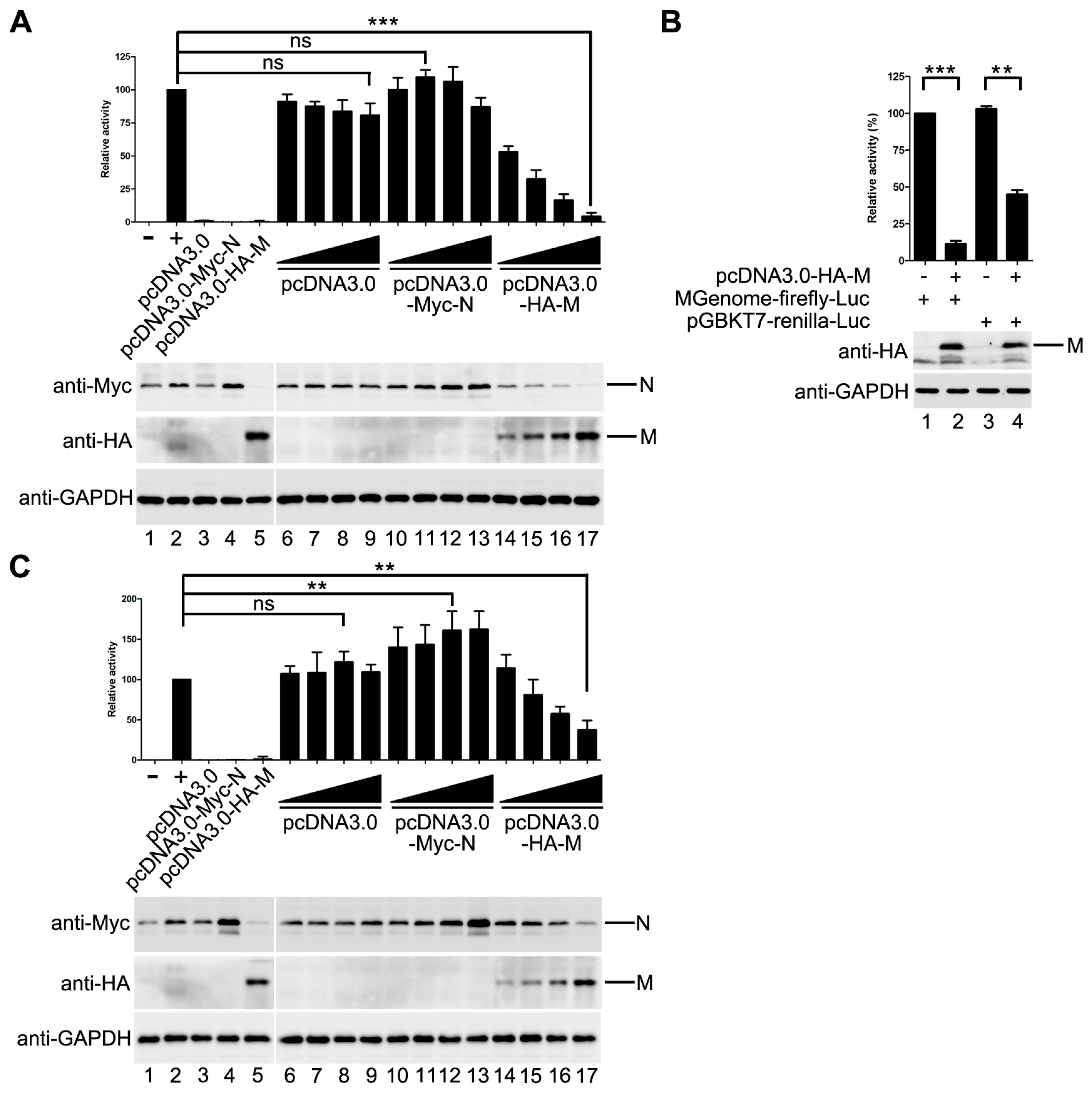
3.1. M Reduces HPIV3 Mingenome-Encoded Reporter Activity
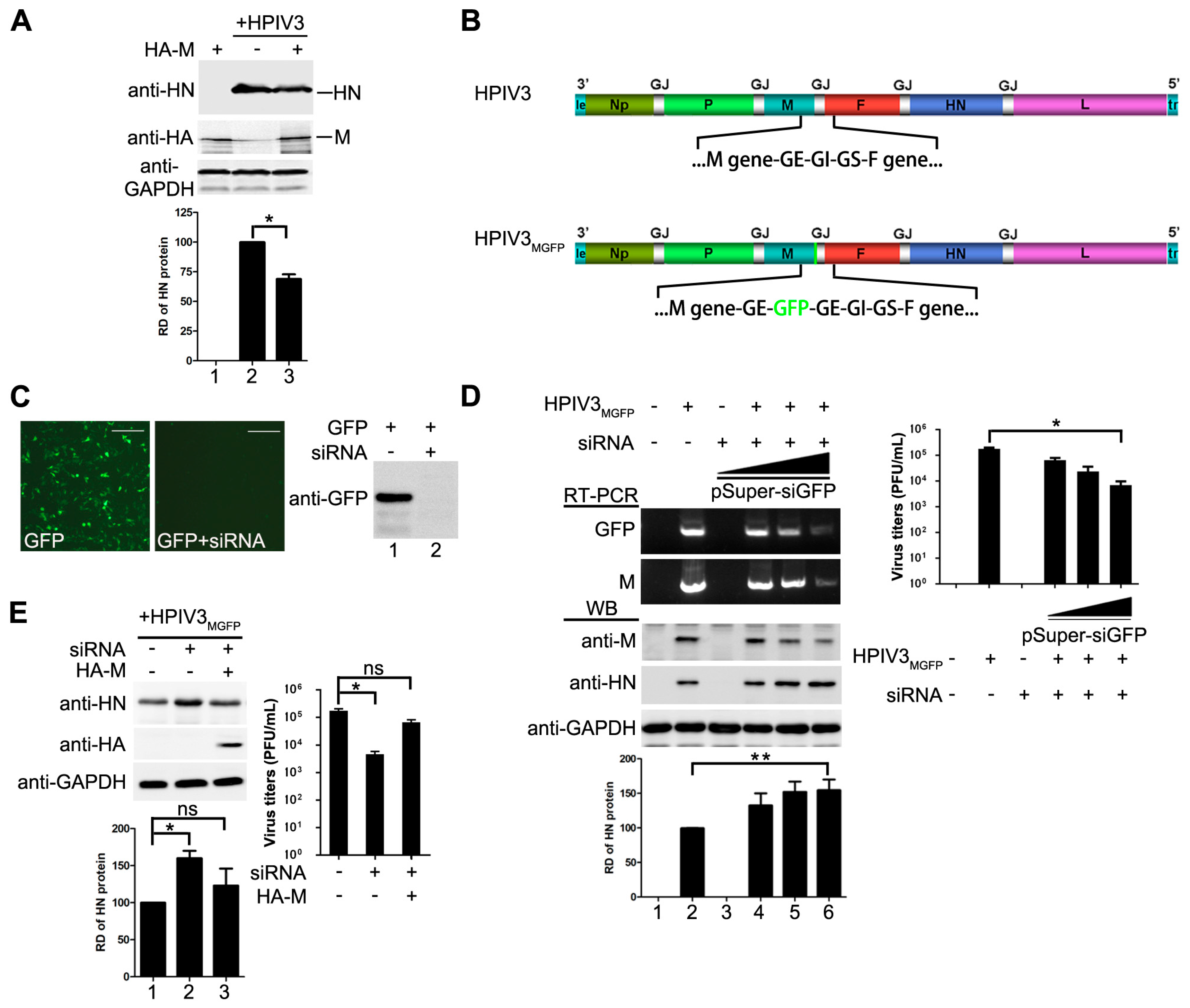
3.2. M Restricts the Replication of HPIV3
3.3. The Inhibitory Activity of M Is Independent of Its Budding Ability
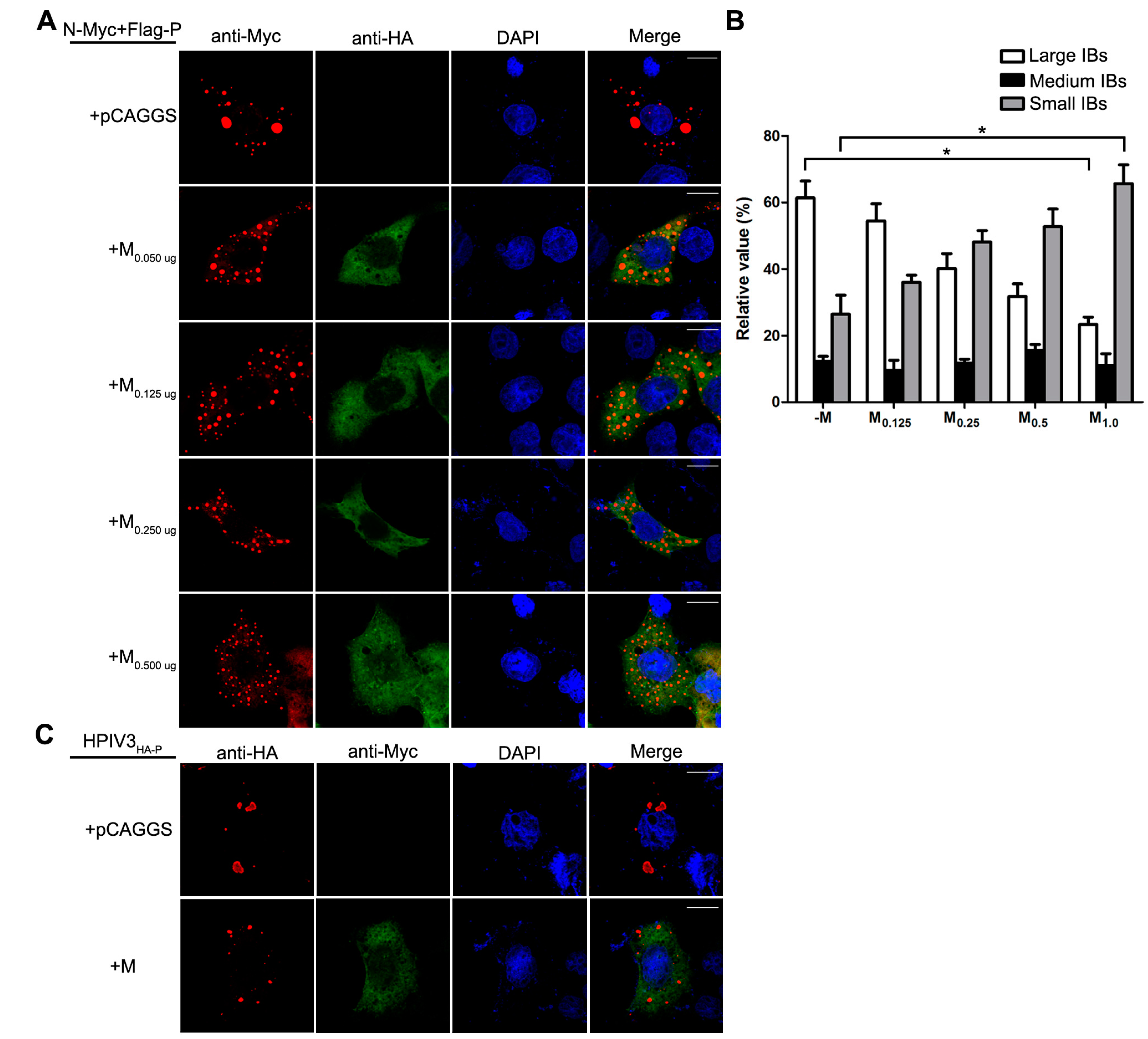
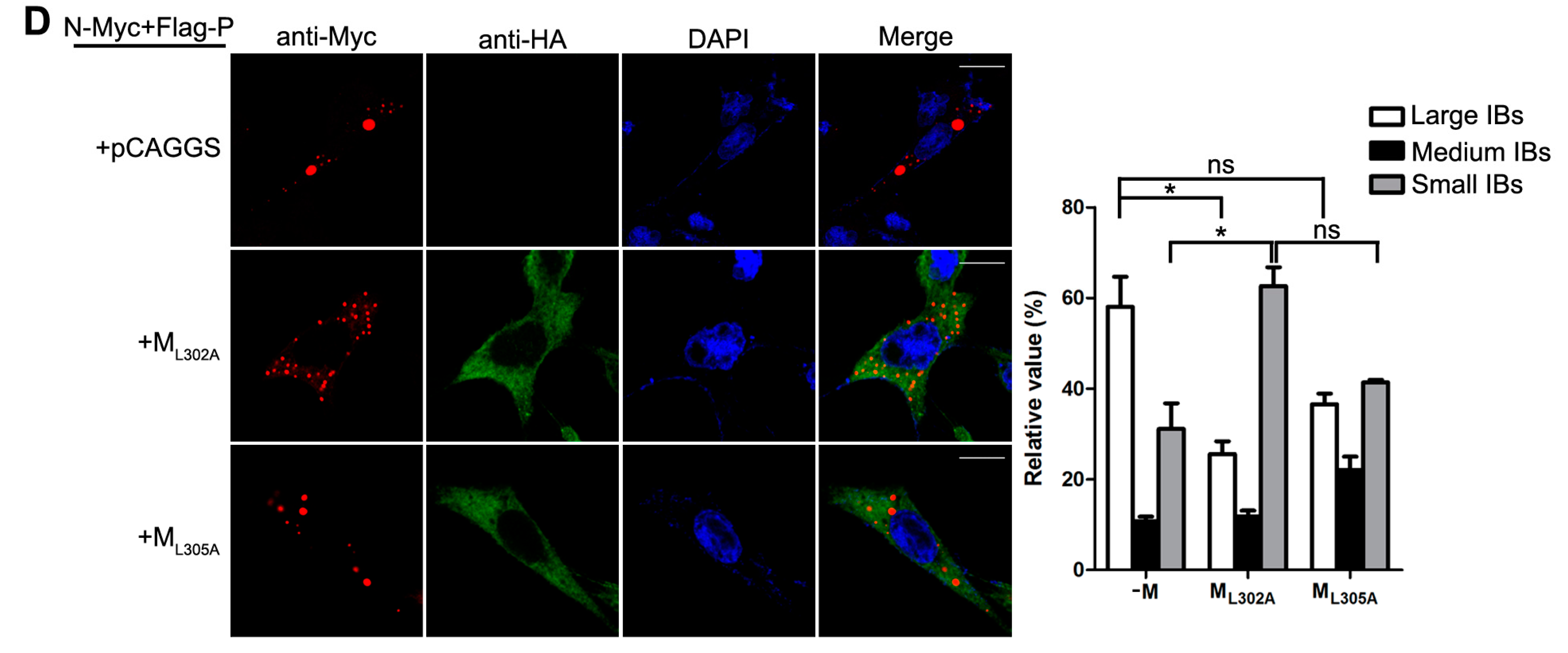
3.4. M Reduces the IB Formation via M–N Interaction
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
Abbreviations
| HPIV3 | human parainfluenza virus type 3 |
| M | matrix protein |
| VLP | virus like particle |
| N | nucleoprotein |
| IBs | inclusion bodies |
| NNSV | non-segmented, negative-strand RNA virus |
| P | phosphoprotein |
| L | RNA-dependent RNA polymerase |
| F | fusion protein |
| HN | hemagglutinin-neuraminidase |
| RNP | ribonucleoprotein |
| GS | gene start |
| GE | gene end |
| GJ | gene junction |
| VSV | vesicular stomatitis virus |
| RSV | respiratory syncytial virus |
| NiV | Nipah virus |
| EBOV | Ebola virus |
| MeV | measles virus |
| PIV5 | parainfluenza virus type 5 |
| IFN-I | type-I interferon |
| RABV | rabies virus |
| MARV | Marburg virus |
| MOI | multiplicity of infection |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | fetal bovine serum |
| vTF7-3 | Recombinant vaccinia virus |
| ORF | Open reading frame |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
References
- Moscona, A. Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease. J. Clin. Investig. 2005, 115, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Barik, S.; De, B.P. Gene expression of nonsegmented negative strand RNA viruses. Pharmacol. Ther. 1991, 51, 47–70. [Google Scholar] [CrossRef]
- Hoffman, M.A.; Banerjee, A.K. Precise mapping of the replication and transcription promoters of human parainfluenza virus type 3. Virology 2000, 269, 201–211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed]
- Myers, T.M.; Pieters, A.; Moyer, S.A. A highly conserved region of the Sendai virus nucleocapsid protein contributes to the NP-NP binding domain. Virology 1997, 229, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, C.J.; Retzler, C.; Homann, H.E.; Neubert, W.J. The carboxy-terminal domain of Sendai virus nucleocapsid protein is involved in complex formation between phosphoprotein and nucleocapsid-like particles. Virology 1994, 204, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.A., Jr.; Favier, A.; Gerard, F.C.; Leyrat, C.; Brutscher, B.; Blondel, D.; Ruigrok, R.W.; Blackledge, M.; Jamin, M. Solution structure of the C-terminal nucleoprotein-RNA binding domain of the vesicular stomatitis virus phosphoprotein. J. Mol. Biol. 2008, 382, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Malur, A.G.; Huo, Y.; De, B.P.; Banerjee, A.K. Characterization of the oligomerization domain of the phosphoprotein of human parainfluenza virus type 3. Virology 2002, 302, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Zhang, G.; Yan, Q.; Yang, X.; Ding, B.; Tang, Q.; Sun, S.; Hu, Z.; Chen, M. An amino acid of human parainfluenza virus type 3 nucleoprotein is critical for template function and cytoplasmic inclusion body formation. J. Virol. 2013, 87, 12457–12470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Jiang, Y.; Cheng, Q.; Zhong, Y.; Qin, Y.; Chen, M. Inclusion Body Fusion of Human Parainfluenza Virus Type 3 Regulated by Acetylated alpha-Tubulin Enhances Viral Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Mebatsion, T.; Weiland, F.; Conzelmann, K.K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 1999, 73, 242–250. [Google Scholar] [PubMed]
- Cathomen, T.; Mrkic, B.; Spehner, D.; Drillien, R.; Naef, R.; Pavlovic, J.; Aguzzi, A.; Billeter, M.A.; Cattaneo, R. A matrix-less measles virus is infectious and elicits extensive cell fusion: Consequences for propagation in the brain. EMBO J. 1998, 17, 3899–3908. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. Viral late domains. J. Virol. 2002, 76, 4679–4687. [Google Scholar] [CrossRef] [PubMed]
- Harty, R.N.; Paragas, J.; Sudol, M.; Palese, P. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: Implications for viral budding. J. Virol. 1999, 73, 2921–2929. [Google Scholar] [PubMed]
- Baviskar, P.S.; Hotard, A.L.; Moore, M.L.; Oomens, A.G. The respiratory syncytial virus fusion protein targets to the perimeter of inclusion bodies and facilitates filament formation by a cytoplasmic tail-dependent mechanism. J. Virol. 2013, 87, 10730–10741. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.R.; Crameri, G.; Wang, L.F.; Eaton, B.T.; Broder, C.C. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol. J. 2007, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Scianimanico, S.; Schoehn, G.; Weissenhorn, W. Vesicular release of ebola virus matrix protein VP40. Virology 2001, 283, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Runkler, N.; Pohl, C.; Schneider-Schaulies, S.; Klenk, H.D.; Maisner, A. Measles virus nucleocapsid transport to the plasma membrane requires stable expression and surface accumulation of the viral matrix protein. Cell Microbiol. 2007, 9, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.P.; Leser, G.P.; Waning, D.L.; Lamb, R.A. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J. Virol. 2002, 76, 3952–3964. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Schmitt, P.T.; Li, Z.; McCrory, T.S.; He, B.; Schmitt, A.P. Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J. Virol. 2009, 83, 7261–7272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, S.; Ding, B.; Yang, X.; Chen, L.; Yan, Q.; Jiang, Y.; Zhong, Y.; Chen, M. A leucine residue in the C terminus of human parainfluenza virus type 3 matrix protein is essential for efficient virus-like particle and virion release. J. Virol. 2014, 88, 13173–13188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, G.; Zhong, Y.; Qin, Y.; Chen, M. Interaction of Human Parainfluenza Virus Type 3 Nucleoprotein with Matrix Protein Mediates Internal Viral Protein Assembly. J. Virol. 2015, 90, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- De, B.P.; Thornton, G.B.; Luk, D.; Banerjee, A.K. Purified matrix protein of vesicular stomatitis virus blocks viral transcription in vitro. Proc. Natl. Acad. Sci. USA 1982, 79, 7137–7141. [Google Scholar] [CrossRef] [PubMed]
- Finke, S.; Mueller-Waldeck, R.; Conzelmann, K.K. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 2003, 84 Pt 6, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Takeda, M.; Shirogane, Y.; Nakatsu, Y.; Nakamura, T.; Yanagi, Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 2009, 83, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Jung, S.; Herwig, A.; Groseth, A.; Becker, S. Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription. Virology 2010, 403, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jans, D.A.; Bardin, P.G.; Meanger, J.; Mills, J.; Ghildyal, R. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J. Virol. 2008, 82, 8863–8870. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Ruth, F.X.; Dessen, A.; Timmins, J.; Bracher, A.; Kolesnikowa, L.; Becker, S.; Klenk, H.D.; Weissenhorn, W. The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties. Structure 2003, 11, 423–433. [Google Scholar] [CrossRef]
- Johnson, R.F.; McCarthy, S.E.; Godlewski, P.J.; Harty, R.N. Ebola virus VP35-VP40 interaction is sufficient for packaging 3E-5E minigenome RNA into virus-like particles. J. Virol. 2006, 80, 5135–5144. [Google Scholar] [CrossRef] [PubMed]
- Neumann, P.; Lieber, D.; Meyer, S.; Dautel, P.; Kerth, A.; Kraus, I.; Garten, W.; Stubbs, M.T. Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc. Natl. Acad. Sci. USA 2009, 106, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Jacamo, R.; Lopez, N.; Wilda, M.; Franze-Fernandez, M.T. Tacaribe virus Z protein interacts with the L polymerase protein to inhibit viral RNA synthesis. J. Virol. 2003, 77, 10383–10393. [Google Scholar] [CrossRef] [PubMed]
- Black, B.L.; Lyles, D.S. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription of target genes in vivo. J. Virol. 1992, 66, 4058–4064. [Google Scholar] [PubMed]
- Wenigenrath, J.; Kolesnikova, L.; Hoenen, T.; Mittler, E.; Becker, S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 2010, 91 Pt 5, 1325–1334. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | VLP Formation | M–N Interaction | M–P Interaction |
|---|---|---|---|
| M | + | + | + |
| ML302A | - | + | + |
| ML305A | + | - | + |
| Category | Size (Mean Cross-Sectional Area) |
|---|---|
| Large | Greater than 8 μm2 |
| Medium | Between 3 μm2 and 8 μm2 |
| Small | Less than 3 μm2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Cheng, Q.; Luo, C.; Qin, Y.; Chen, M. Human Parainfluenza Virus Type 3 Matrix Protein Reduces Viral RNA Synthesis of HPIV3 by Regulating Inclusion Body Formation. Viruses 2018, 10, 125. https://doi.org/10.3390/v10030125
Zhang S, Cheng Q, Luo C, Qin Y, Chen M. Human Parainfluenza Virus Type 3 Matrix Protein Reduces Viral RNA Synthesis of HPIV3 by Regulating Inclusion Body Formation. Viruses. 2018; 10(3):125. https://doi.org/10.3390/v10030125
Chicago/Turabian StyleZhang, Shengwei, Qi Cheng, Chenxi Luo, Yali Qin, and Mingzhou Chen. 2018. "Human Parainfluenza Virus Type 3 Matrix Protein Reduces Viral RNA Synthesis of HPIV3 by Regulating Inclusion Body Formation" Viruses 10, no. 3: 125. https://doi.org/10.3390/v10030125
APA StyleZhang, S., Cheng, Q., Luo, C., Qin, Y., & Chen, M. (2018). Human Parainfluenza Virus Type 3 Matrix Protein Reduces Viral RNA Synthesis of HPIV3 by Regulating Inclusion Body Formation. Viruses, 10(3), 125. https://doi.org/10.3390/v10030125