The Bacteriophage Lambda CII Phenotypes for Complementation, Cellular Toxicity and Replication Inhibition Are Suppressed in cII-oop Constructs Expressing the Small RNA OOP


Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Construction, Bacterial and Phage Strains Employed
2.2. Fluorescence Assays for Expressed D-GFPuv and GFPuv*
2.3. Complementation Assay for CII
2.4. Determination of Toxicity and Plasmid Loss
2.5. Oligonucleotides Used for Plasmid Construction and Sequencing
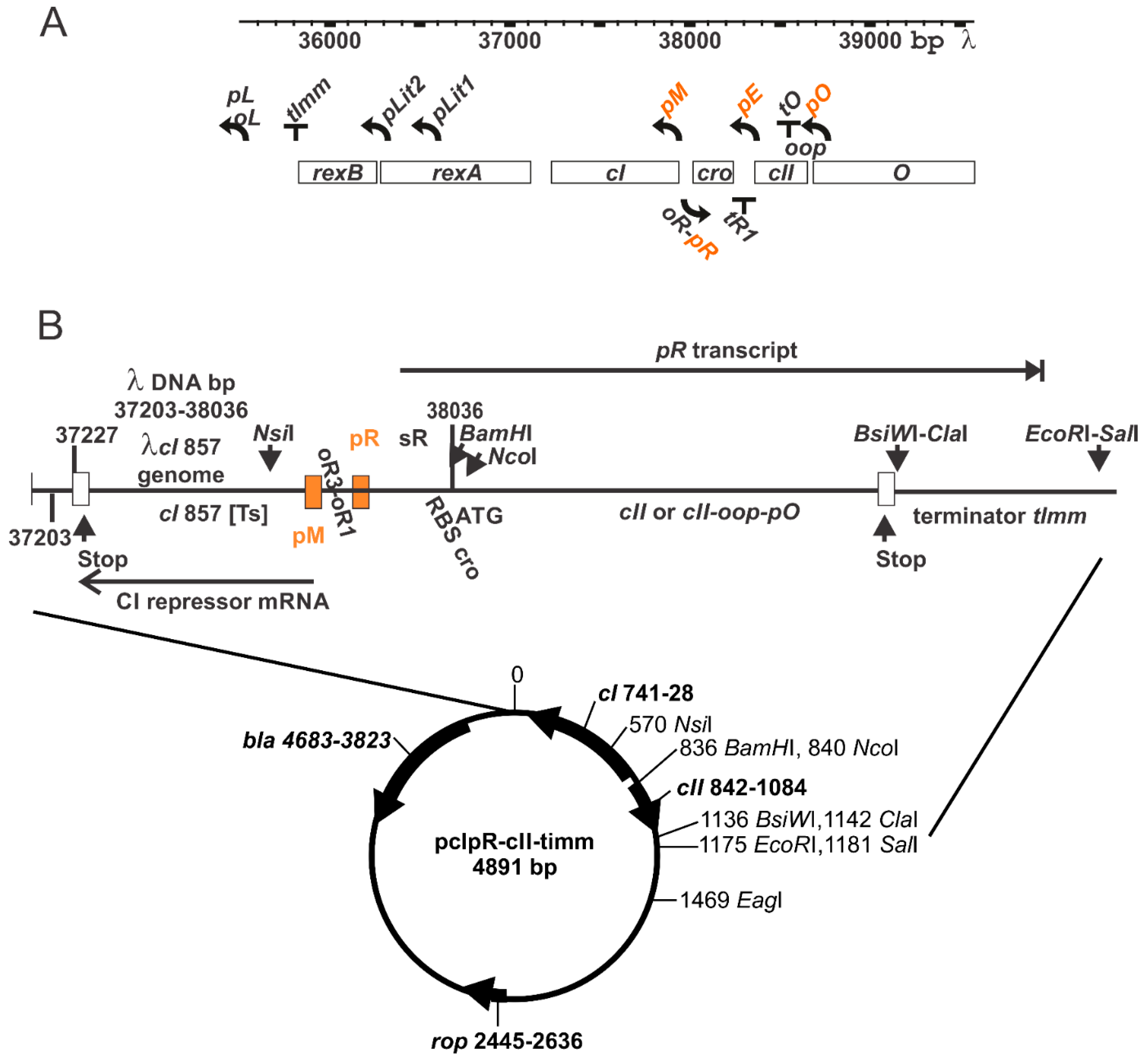
2.6. Gene Expression Plasmid
3. Results
3.1. Thermally Inducible Gene Expression from pcIpR[]Timm Plasmid
3.2. Untangling CII Activities: Cellular Toxicity and Promoting Plasmid Loss
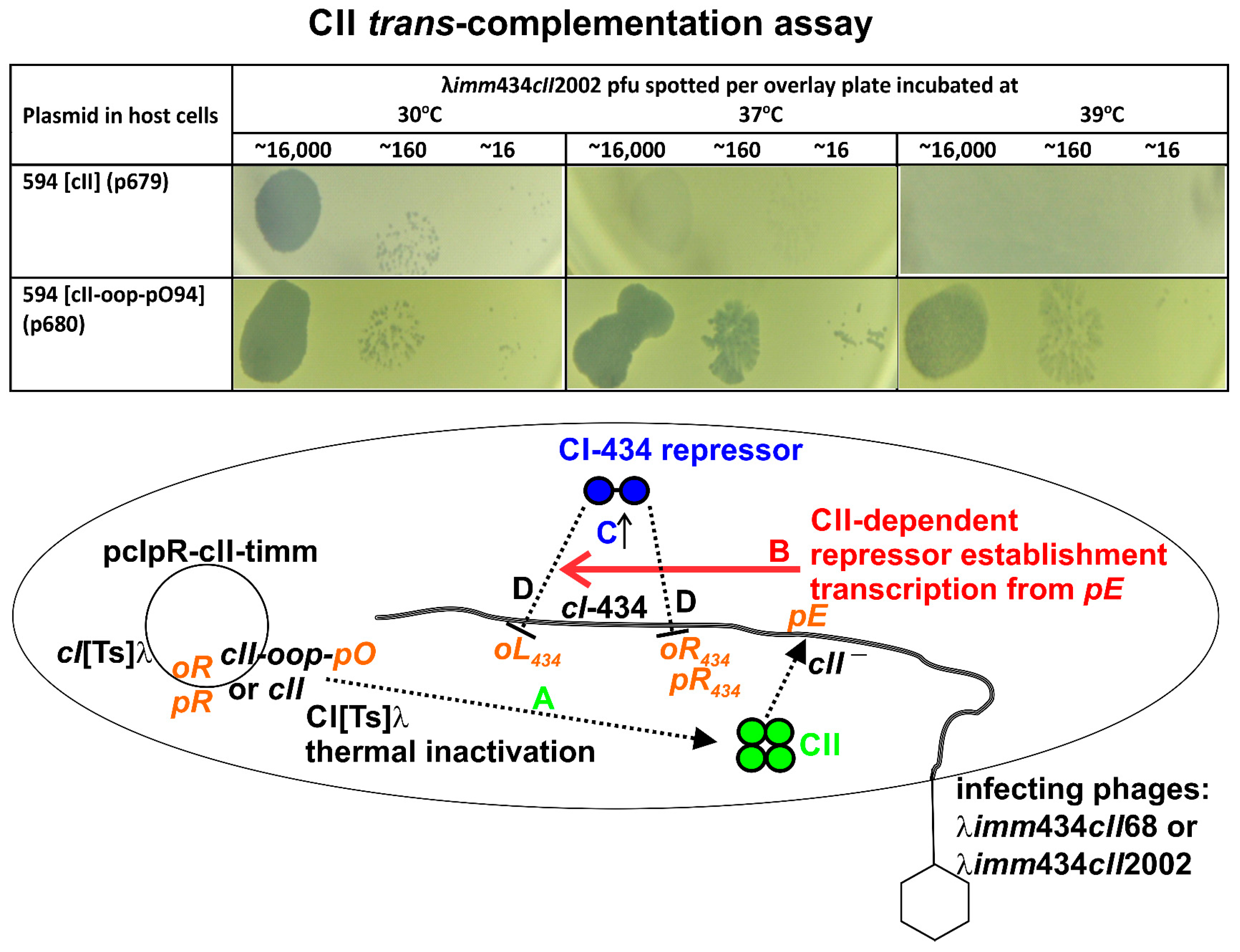
3.3. CII Complementation
3.4. Host Modulation of CII-Dependent Cellular Toxicity and Plasmid Retention
3.5. Influence of oop RNA Expression on cII-Dependent Cellular Toxicity and Plasmid Loss
3.6. Influence of Terminally Overlapping Divergent cII and oop Transcription on CII Complementation
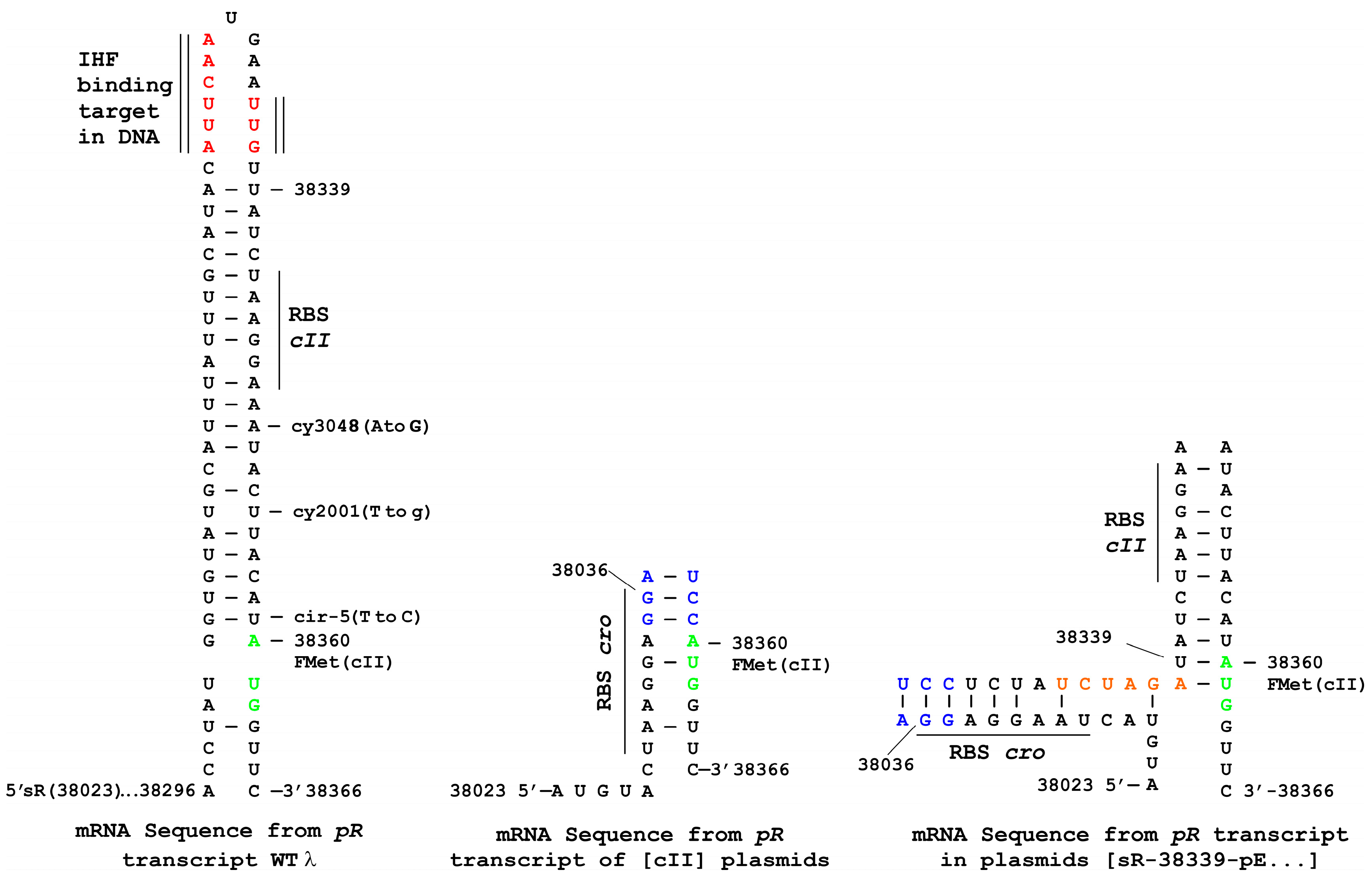
3.7. Plasmid Construction Variations and Influence on cII Expression
4. Discussion
5. Conclusions
- The results confirm that OOP RNA expression from the genetic element pO-oop-to can suppress high CII activity and that OOP RNA likely serves as a powerful regulatory pivot in temperate lambdoid phage development.
- Plasmids with a pO94, comprising 94 bases rightward from oop, prevented CII complementation, CII-dependent plasmid loss and suppressed CII toxicity, suggesting that the active pO promoter to produce OOP requires an extended DNA sequence, beyond that required to encode the −10 and −35 regions.
- All three CII activities were eliminated by the deletion of its COOH-terminal 20 amino acids.
- E. coli mutations were shown to influence CII activities. (a) Inactivating the hflA locus encoding HflK-HflC proteins that modulate the FtsH ATP-dependent membrane protease significantly reduced CII trans-complementation and toxicity; (b) A null allele of pcnB, encoding poly (A) polymerase I, eliminated CII complementation and increased CII toxicity; (c) Five of six rpoB point mutations significantly reduced CII trans-complementation; (d) The CII1–87 mutant, deleted for the terminal 10 amino acids, lost its ability to complement in five rpoB mutant cells.
- The results suggest that the terminal end of CII likely interacts with the β-subunit of RNA polymerase.
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Court, D.L.; Oppenheim, A.B.; Adhya, S.L. A new look at bacteriophage lambda genetic networks. J. Bacteriol. 2007, 189, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.; Casjens, S. Bacteriophage lambda and its genetic neighborhood. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 409–447. [Google Scholar]
- Dodd, I.B.; Shearwin, K.E.; Egan, J.B. Revisited gene regulation in bacteriophage lambda. Curr. Opin. Genet. Dev. 2005, 15, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, I.; Hagen, D. The lysis-lysogeny decision of phage lambda: Explicit programming and responsiveness. Annu. Rev. Genet. 1980, 14, 399–445. [Google Scholar] [CrossRef] [PubMed]
- Little, J.W. Lysogeny, prophage induction and lysogenic conversion. In Phages: Their Role in Bacterial Pathogenesis and Biotechnology; Waldor, M.K., Friedman, D.I., Adhya, S.L., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 37–54. [Google Scholar]
- Kobiler, O.; Oppenheim, A.B.; Herman, C. Recruitment of host ATP-dependent proteases by bacteriophage lambda. J. Struct. Biol. 2004, 146, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Rokney, A.; Friedman, N.; Court, D.L.; Stavans, J.; Oppenheim, A.B. Quantitative kinetic analysis of the bacteriophage lambda genetic network. Proc. Natl. Acad. Sci. USA 2005, 102, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.B.; Kobiler, O.; Stavans, J.; Court, D.L.; Adhya, S. Switches in bacteriophage lambda development. Annu. Rev. Genet. 2005, 39, 409–429. [Google Scholar] [CrossRef] [PubMed]
- Herman, C.; Thevenet, D.; D’Ari, R.; Bouloc, P. The HflB protease of Escherichia coli degrades its inhibitor lambda cIII. J. Bacteriol. 1997, 179, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Akiyama, Y.; Ito, K. Host regulation of lysogenic decision in bacteriophage lambda: Transmembrane modulation of ftsh (HflB), the cii degrading protease, by hflkc (HflA). Proc. Natl. Acad. Sci. USA 1997, 94, 5544–5549. [Google Scholar] [CrossRef] [PubMed]
- Shotland, Y.; Koby, S.; Teff, D.; Mansur, N.; Oren, D.A.; Tatematsu, K.; Tomoyasu, T.; Kessel, M.; Bukau, B.; Ogura, T.; et al. Proteolysis of the phage lambda cII regulatory protein by ftsh (HflB) of Escherichia coli. Mol. Microbiol. 1997, 24, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Kihara, A.; Akiyama, Y.; Ito, K. A protease complex in the escherichia coli plasma membrane: Hflkc (HflA) forms a complex with ftsh (HflB), regulating its proteolytic activity against secy. EMBO J. 1996, 15, 6122–6131. [Google Scholar] [PubMed]
- Kihara, A.; Akiyama, Y.; Ito, K. Revisiting the lysogenization control of bacteriophage lambda. Identification and characterization of a new host component, HflD. J. Biol. Chem. 2001, 276, 13695–13700. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Koby, S.; Teff, D.; Court, D.; Oppenheim, A.B. The phage lambda cII transcriptional activator carries a C-terminal domain signaling for rapid proteolysis. Proc. Natl. Acad. Sci. USA 2002, 99, 14964–14969. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, K.; Parua, P.K.; Datta, A.B.; Parrack, P. Escherichia coli HflK and HflC can individually inhibit the HflB (ftsh)-mediated proteolysis of lambdacii in vitro. Arch. Biochem. Biophys. 2010, 501, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Rokney, A.; Oppenheim, A.B. Phage lambda cIII: A protease inhibitor regulating the lysis-lysogeny decision. PLoS ONE 2007, 2, e363. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, M.A.; Knight, D.M.; Das, A.; Miller, H.I.; Echols, H. Control of phage lambda development by stability and synthesis of cII protein: Role of the viral cIII and host HFLA, HIMA and HIMD genes. Cell 1982, 31, 565–573. [Google Scholar] [CrossRef]
- Krinke, L.; Wulff, D.L. Oop RNA, produced from multicopy plasmids, inhibits lambda cII gene expression through an rnase III-dependent mechanism. Genes Dev. 1987, 1, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Krinke, L.; Wulff, D.L. Rnase III-dependent hydrolysis of lambda cII-O gene mRNA mediated by lambda oop antisense RNA. Genes Dev. 1990, 4, 2223–2233. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Szybalski, W. Control of short leftward transcripts from the immunity and ORI regions in induced coliphage lambda. Mol. Gen. Genet. 1973, 126, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Hayes, C. Control of lambda repressor prophage and establishment transcription by the product of gene TOF. Mol. Gen. Genet. 1978, 164, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Hayes, C. Control of bacteriophage lambda repressor establishment transcription: Kinetics of l-strand transcription from the y-cII-oop-O-P region. Mol. Gen. Genet. 1979, 170, 75–88. [Google Scholar] [PubMed]
- Hayes, S.; Slavcev, R.A. Polarity within pM and pE promoted phage lambda cI-rexA-rexB transcription and its suppression. Can. J. Microbiol. 2005, 51, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, L.; Kaiser, A.D. Control of lambda repressor synthesis. Proc. Natl. Acad. Sci. USA 1971, 68, 2185–2189. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S. Phage Lambda Display Constructions. U.S. Patent 8663913 b 1, 4 March 2014. [Google Scholar]
- Hayes, S. Phage Lambda Display Constructions. Canadian Patent 2761105, 10 February 2014. [Google Scholar]
- Hayes, S.; Gamage, L.N.; Hayes, C. Dual expression system for assembling phage lambda display particle (LDP) vaccine to porcine circovirus 2 (PCV2). Vaccine 2010, 28, 6789–6799. [Google Scholar] [CrossRef] [PubMed]
- Simatake, H.; Rosenberg, M. Purified lambda regulatory protein cII positively activates promoters for lysogenic development. Nature 1981, 292, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, B.; Glinkowska, M.; Iwanicki, A.; Obuchowski, M.; Sojka, P.; Thomas, M.S.; Wegrzyn, G. Toxicity of the bacteriophage lambda cII gene product to Escherichia coli arises from inhibition of host cell DNA replication. Virology 2003, 313, 622–628. [Google Scholar] [CrossRef]
- Kourilsky, P.; Gros, D. Lysogenization by bacteriophage lambda IV inhibition of phage DNA synthesis by the products of genes cII and cIII. Biochimie 1976, 58, 1321–1327. [Google Scholar] [CrossRef]
- McMacken, R.; Mantei, N.; Butler, B.; Joyner, A.; Echols, H. Effect of mutations in the C2 and C3 genes of bacteriophage lambda on macromolecular synthesis in infected cells. J. Mol. Biol. 1970, 49, 639–655. [Google Scholar] [CrossRef]
- Szalewska-Palasz, A.; Wrobel, B.; Wegrzyn, G. Rapid degradation of polyadenylated oop RNA. FEBS Lett. 1998, 432, 70–72. [Google Scholar] [CrossRef]
- Wrobel, B.; Herman-Antosiewicz, A.; Szalewska-Palasz, S.; Wegrzyn, G. Polyadenylation of oop RNA in the regulation of bacteriophage lambda development. Gene 1998, 212, 57–65. [Google Scholar] [CrossRef]
- Wulff, D.L.; Rosenberg, M.R. Establishment of repressor synthesis. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F.W., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1983; pp. 519–684. [Google Scholar]
- Hayes, S.; Wang, W.; Rajamanickam, K.; Chu, A.; Banerjee, A.; Hayes, C. Lambda gpP-DnaB helicase sequestration and gpP-RpoB associated effects: On screens for auxotrophs, selection for Rif(R), toxicity, mutagenicity, plasmid curing. Viruses 2016, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage lambda P protein: Trans-activation, inhibition phenotypes and their suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Horbay, M.A.; Hayes, C. A CI-independent form of replicative inhibition: Turn off of early replication of bacteriophage lambda. PLoS ONE 2012, 7, e36498. [Google Scholar] [CrossRef] [PubMed]
- Krinke, L.; Mahoney, M.; Wulff, D.L. The role of the oop antisense RNA in Coliphage lambda development. Mol. Microbiol. 1991, 5, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Schroeder, J.L.; Szybalski, W.; Sanger, F.; Blattner, F.R. Appendix I. A molecular map of coliphage lambda. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: New York, NY, USA, 1983; pp. 469–517. [Google Scholar]
- Dodd, I.B.; Perkins, A.J.; Tsemitsidis, D.; Egan, J.B. Octamerization of lambda CI repressor is needed for effective repression of P(RM) and efficient switching from lysogeny. Genes Dev. 2001, 15, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Dodd, I.B.; Shearwin, K.E.; Perkins, A.J.; Burr, T.; Hochschild, A.; Egan, J.B. Cooperativity in long-range gene regulation by the lambda cI repressor. Genes Dev. 2004, 18, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Revet, B.; von Wilcken-Bergmann, B.; Bessert, H.; Barker, A.; Muller-Hill, B. Four dimers of lambda repressor bound to two suitably spaced pairs of lambda operators form octamers and DNA loops over large distances. Curr. Biol. 1999, 9, 151–154. [Google Scholar] [CrossRef]
- Svenningsen, S.L.; Costantino, N.; Court, D.L.; Adhya, S. On the role of CRO in lambda prophage induction. Proc. Natl. Acad. Sci. USA 2005, 102, 4465–4469. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.; Weissbach, H.; Nash, H.A. In vitro regulation of phage lambda cII gene expression by Escherichia coli integration host factor. Proc. Natl. Acad. Sci. USA 1984, 81, 6009–6013. [Google Scholar] [CrossRef] [PubMed]
- Mahajna, J.; Oppenheim, A.B.; Rattray, A.; Gottesman, M. Translation initiation of bacteriophage lambda gene cII requires integration host factor. J. Bacteriol. 1986, 165, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Schroeder, J.L.; Szybalski, W.; Sanger, F.; Coulson, A.R.; Hong, G.F.; Hill, D.F.; Petersen, G.B.; Blattner, F.R. Appendix II. Complete annotated lambda sequence. In Lambda II; Hendrix, R.W., Roberts, J.W., Stahl, F., Weisberg, R.A., Eds.; Cold Spring Harbor Laboratory: New York, NY, USA, 1983; pp. 519–676. [Google Scholar]
- Nunez, J.K.; Bai, L.; Harrington, L.B.; Hinder, T.L.; Doudna, J.A. CRISPR immunological memory requires a host factor for specificity. Mol. Cell 2016, 62, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Grosschedl, R.; Schwarz, E. Nucleotide sequence of the cro-cII-oop region of bacteriophage 434 DNA. Nucleic Acids Res. 1979, 6, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Jasiecki, J.; Wegrzyn, G. Growth-rate dependent RNA polyadenylation in Escherichia coli. EMBO Rep. 2003, 4, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Scherer, G.; Hobom, G.; Kossel, H. DNA base sequence of the Po promoter region of phage lamdba. Nature 1977, 265, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.; Court, D.; Shimatake, H.; Brady, C.; Wulff, D.L. The relationship between function and DNA sequence in an intercistronic regulatory region in phage lambda. Nature 1978, 272, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Belfort, M.; Wulff, D.L. Genetic and biochemical investigation of the Escherichia coli mutant hfl-1 which is lysogenized at high frequency by bacteriophage lambda. J. Bacteriol. 1973, 115, 299–306. [Google Scholar] [PubMed]
- Hammer, K.; Jensen, K.F.; Poulsen, P.; Oppenheim, A.B.; Gottesman, M. Isolation of Escherichia coli rpob mutants resistant to killing by lambda cII protein and altered in pyre gene attenuation. J. Bacteriol. 1987, 169, 5289–5297. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Mahoney, M.E.; Wulff, D.L.; Rosenberg, M. Identification of the DNA binding domain of the phage lambda cII transcriptional activator and the direct correlation of CII protein stability with its oligomeric forms. Genes Dev. 1988, 2, 184–195. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, M.S.; Glass, R.E. Escherichia coli RPOA mutation which impairs transcription of positively regulated systems. Mol. Microbiol. 1991, 5, 2719–2725. [Google Scholar] [CrossRef] [PubMed]
- Obuchowski, M.; Giladi, H.; Koby, S.; Szalewska-Palasz, A.; Wegrzyn, A.; Oppenheim, A.B.; Thomas, M.S.; Wegrzyn, G. Impaired lysogenisation of the Escherichia coli rpoa341 mutant by bacteriophage lambda is due to the inability of cII to act as a transcriptional activator. Mol. Gen. Genet. 1997, 254, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Giffard, P.M.; Booth, I.R. The rpoa341 allele of Escherichia coli specifically impairs the transcription of a group of positively-regulated operons. Mol. Gen. Genet. 1988, 214, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Hollatz, I. Coliphage lambda to terminator lowers the stability of messenger RNA in Escherichia coli hosts. Gene 1988, 72, 119–128. [Google Scholar] [CrossRef]
- Ingle, C.A.; Kushner, S.R. Development of an in vitro mRNA decay system for Escherichia coli: Poly(A) polymerase I is necessary to trigger degradation. Proc. Natl. Acad. Sci. USA 1996, 93, 12926–12931. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, N. Polyadenylation of mRNA in bacteria. Microbiology 1996, 142 Pt 11, 3125–3133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, L.K.; Harlow, G.R.; Gregg-Jolly, L.A.; Mount, D.W. Identification of high affinity binding sites for lexa which define new DNA damage-inducible genes in Escherichia coli. J. Mol. Biol. 1994, 241, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S. Initiation of coliphage lambda replication, lit, oop rna synthesis and effect of gene dosage on transcription from promoters PL, PR and PR. Virology 1979, 97, 415–438. [Google Scholar] [CrossRef]
- Oppenheim, A.B.; Rattray, A.J.; Bubunenko, M.; Thomason, L.C.; Court, D.L. In vivo recombineering of bacteriophage lambda by PCR fragments and single-strand oligonucleotides. Virology 2004, 319, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Skinner, S.O.; Zong, C.; Sippy, J.; Feiss, M.; Golding, I. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell 2010, 141, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Schlosser-Silverman, E.; Elgrably-Weiss, M.; Rosenshine, I.; Kohen, R.; Altuvia, S. Characterization of Escherichia coli DNA lesions generated within j774 macrophages. J. Bacteriol. 2000, 182, 5225–5230. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Induction Time (min) | Culture up-Shift from 30 °C ± SE a | ||
|---|---|---|---|
| 37 °C | 39 °C | 42 °C | |
| 0 | 0 (0.2) | 0 (4.0) | 3.2 (0.05) |
| 20 | 0 (0.2) | 1.4 (2.6) | 153.8 (0.5) |
| 40 | 0 (0.3) | 6.7 (2.0) | 415.4 (0.4) |
| 60 | 0.8 (0.3) | 23.2 (3.2) | 518.5 (0.5) |
| 90 | 0 (0.3) | 41.3 (0.3) | 624.6 (0.4) |
| 120 | 7.3 (14.9) | 30.0 (2.9) | 721.2 (0.4) |
| 150 | 10.8 (1.3) | 84.3 (3.4) | 808.4 (0.4) |
| 180 | 23.9 (2.6) | 136.9 (10.7) | 1039.5 (0.7) |
| Host [Plasmid] a | Intensity of CII-Activated CI434 Repression at 37–39 °C b | λimm434 cII− Plaque Formation at 37–39 °C c | Cell Viability, (±SE) and [% Plasmid Loss] Per Growth Temp. of Transformants e | |
|---|---|---|---|---|
| 39 °C | 42 °C | |||
| 594 [cII] = [cII1–97] | H (high) | − | 0.03 (0.001) [0] | <0.001 (0.0001) [100] |
| 594 [cII-oop-pO94] | 0 (none) | + d | 0.59 (0.03) [0] | 0.08 (0.03) [0] |
| 594 [cII1–92] | H | − | 0.73 (0.01) [0] | 0.005 (0.001) [100] |
| 594 [cII1–87] | H | − | 0.76 (0.02) [0] | 0.08 (0.03) [0] |
| 594 [cII1–77] | 0 | + d | 0.74 (0.10) [0] | 0.32 (0.10) [0] |
| 594 [cII-3638](38,642: A-G, M-V) | H | − | 0.62 (0.10) [0] | 0.002 (0.0004) [89] |
| 594 [cII-3639](38,634: A-G, Q-R) | H | − | 0.76 (0.02) [0] | <0.001 (<0.0001) [19] |
| 594 [cII-3638–3639] | H | − | 0.65 (0.04) [0] | <0.001 (<0.0001) [6] |
| 594 hflA::kan [cII] | S (slight) | + | 0.89 (0.01) [0] | 0.008 (<0.0001) [100] |
| 594 hflA::kan [cII1–92] | S | + | 0.69 (0.01) [0] | <0.001 (<0.0001) [100] |
| 594 hflA::kan [cII1–87] | S | + | 0.85 (0.02) [0] | 0.24 (<0.0001) [0] |
| 594 hflA::kan [cII1–77] | 0 | + d | 0.86 (0.10) [0] | 0.58 (0.01) [0] |
| 594 hflA::kan [cII-oop-pO94] | 0 | + d | 0.63 (<0.0001) [0] | 0.001 (0.0002) [100] |
| 594 pcnB::kan [cII] | 0 | + d | <0.001 (0.01) [67] | <0.001 (0.0001) [100] |
| 594 pcnB::kan [cII-oop-pO94] | 0 | + d | 0.46 (0.01) [64] | <0.001 (0.0003) [89] |
| Host(s) Strains a | CII-Construct Plasmid(s) with Thermally Inducible Expression of cII Allele | Intensity of CII-Activated CI434 Repression at 37–39 °C b | λimm434 cII− Plaque Formation at 37–39 °C b |
|---|---|---|---|
| 594 rpoB B1 | cII; cII1–92 | H | − |
| 594 rpoB B8, C1, C4, C10, D2, D6 | cII; cII1–92 | S | + |
| 594 rpoB B1, C1 | cII1–87 | S | + |
| 594 rpoB B8, C4, C10, D2, D6 | cII1–87 | 0 | + c |
| Host Strains with rpoB Alleles | Cell Viability (±SE) [% Plasmid Loss] Per Growth of Transformant at 42 °C | |
|---|---|---|
| [cII] a | [cII-oop-pO94] b | |
| 594 c | <0.001 (0.001) [100] | 0.08 (0.03) [0] |
| 594 rpoB B1 | <0.001 (<0.0001) [100] | 0.07 (0.01) [0] |
| 594 rpoB B8 | 0.01 (<0.0001) [78] | 0.13 (0.03) [0] |
| 594 rpoB C1 | 0.007 (0.10) [100] | 0.36 (<0.0001) [0] |
| 594 rpoB C4 | 0.003 (0.010) [0] | 0.07 (<0.0001) [0] |
| 594 rpoB C10 | 0.004 (0.001) [100] | 1.00 (0.01) [0] |
| 594 rpoB D2 | 0.38 (<0.0001) [0] | 0.10 (<0.0001) [81] |
| 594 rpoB D6 | 0.02 (0.01) [6] | 0.03 (0.01) [86] |
| CII-Construct Plasmids with Thermally Inducible Expression of cII Allele | Intensity of CII-Activated CI434 Expression at 37–39 °C a | λimm434 cII− Plaque Formation at 37–39 °C a | Cell Viability (±SE) [% Plasmid Loss] Per Growth Temp. of Transformants | |
|---|---|---|---|---|
| 39 °C | 42 °C | |||
| sR-38339-pE-cII (p747) | H | − | 0.62 (0.10) [0] | <0.001 (<0.004) [94] |
| sR-38339-pE-cII-oop-pO94 (p748) | 0 | + d | 0.82 (0.02) [0] | 0.001 (<0.0001) [94] |
| sR-38339-pE-cII-cy3048 in −10 pE (p767) | 0-S b | + | 0.89 (0.01) [0] | 0.08 (<0.0001) [0] |
| sR-38339-pE-cII-cy2001 in −10 pE (p765) | 0-S b | + | 0.69 (<0.0001) [0] | 0.01 (<0.0001) [0] |
| sR-38339-pE-cII-cy42 in −35 pE (p764) | 0-S b | + | 1.00 (0.020) [0] | 1.00 (<0.0001) [42] |
| sR-38339-pE-cII-cy3001 in −35 pE (p766) | H | − | 0.72 (<0.0001) [0] | <0.001 (0.001) [8] |
| cII-oop-pO45WT (p763) | H | − | 0.72 (0.10) [0] | 0.07 (<0.0001) [0] |
| cII-oop-pO45-38,683-87MH2 in −10 pO (p759) | H | − | 0.67 (0.10) [0] | <0.001 (0.002) [100] |
| cII-oop-pO45-38683LK in −10 pO (p762) | H | − | 0.78 (0.20) [0] | <0.001 (<0.0001) [50] |
| cII-oop-pO94-38683-87MH (p681) c | H | − | 0.71 (<0.0001) [0] | <0.001 (<0.0001) [86] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajamanickam, K.; Hayes, S. The Bacteriophage Lambda CII Phenotypes for Complementation, Cellular Toxicity and Replication Inhibition Are Suppressed in cII-oop Constructs Expressing the Small RNA OOP. Viruses 2018, 10, 115. https://doi.org/10.3390/v10030115
Rajamanickam K, Hayes S. The Bacteriophage Lambda CII Phenotypes for Complementation, Cellular Toxicity and Replication Inhibition Are Suppressed in cII-oop Constructs Expressing the Small RNA OOP. Viruses. 2018; 10(3):115. https://doi.org/10.3390/v10030115
Chicago/Turabian StyleRajamanickam, Karthic, and Sidney Hayes. 2018. "The Bacteriophage Lambda CII Phenotypes for Complementation, Cellular Toxicity and Replication Inhibition Are Suppressed in cII-oop Constructs Expressing the Small RNA OOP" Viruses 10, no. 3: 115. https://doi.org/10.3390/v10030115
APA StyleRajamanickam, K., & Hayes, S. (2018). The Bacteriophage Lambda CII Phenotypes for Complementation, Cellular Toxicity and Replication Inhibition Are Suppressed in cII-oop Constructs Expressing the Small RNA OOP. Viruses, 10(3), 115. https://doi.org/10.3390/v10030115