The Application of NHEJ-CRISPR/Cas9 and Cre-Lox System in the Generation of Bivalent Duck Enteritis Virus Vaccine against Avian Influenza Virus



 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cells and Transfection
2.2. Virus Infection and Titration
2.3. Multi-Step Growth Curve
2.4. Immunochemistry
2.5. Western Blot
2.6. DEV Genome Extraction and High-Resolution Melting (HRM)
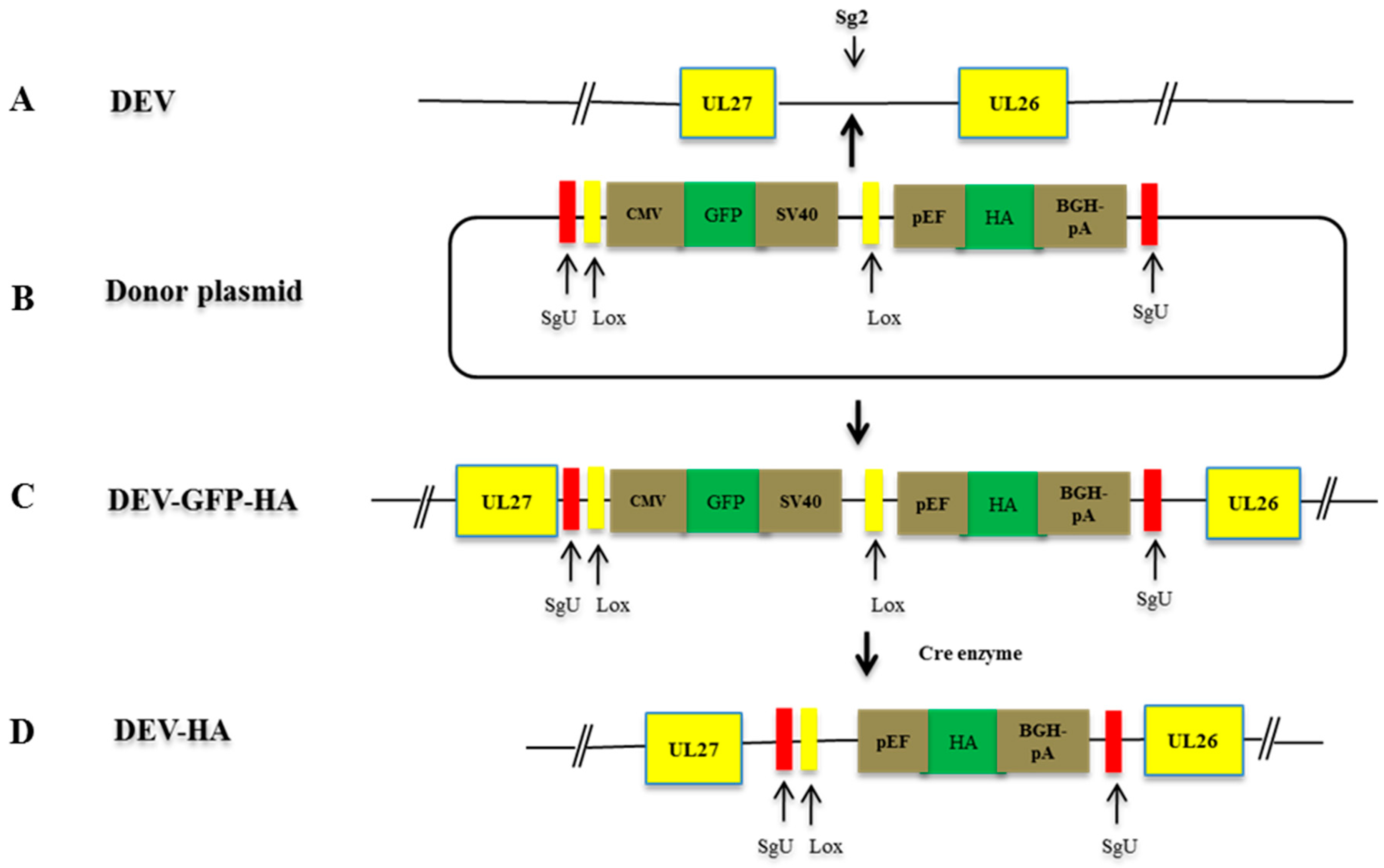
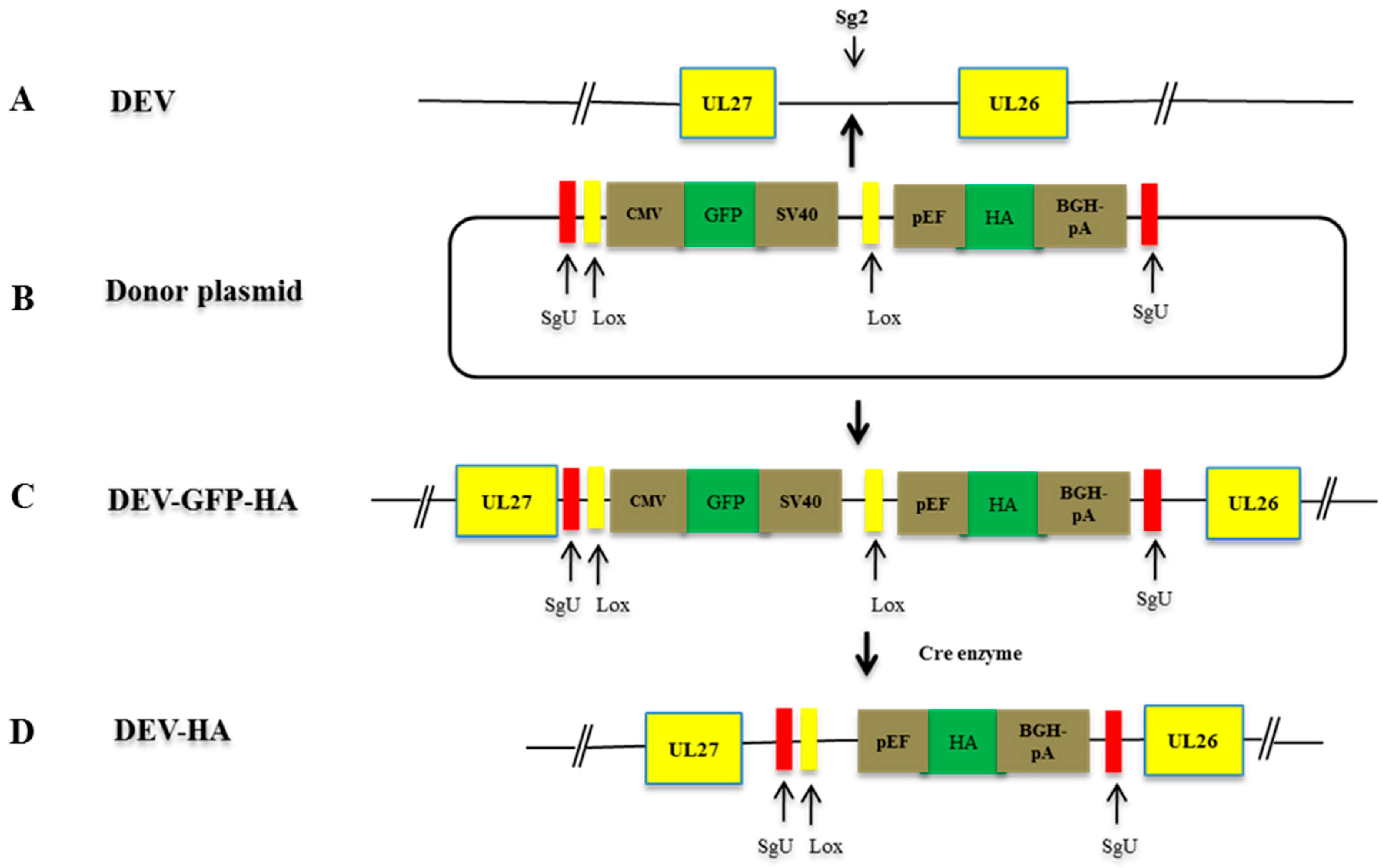
2.7. Construction of sgRNAs and Donor Plasmids
2.8. NHEJ-CRISPR/Cas9-Mediated Gene Insertion
2.9. Cre Enzyme Treatment
2.10. Statistical Analysis
3. Results
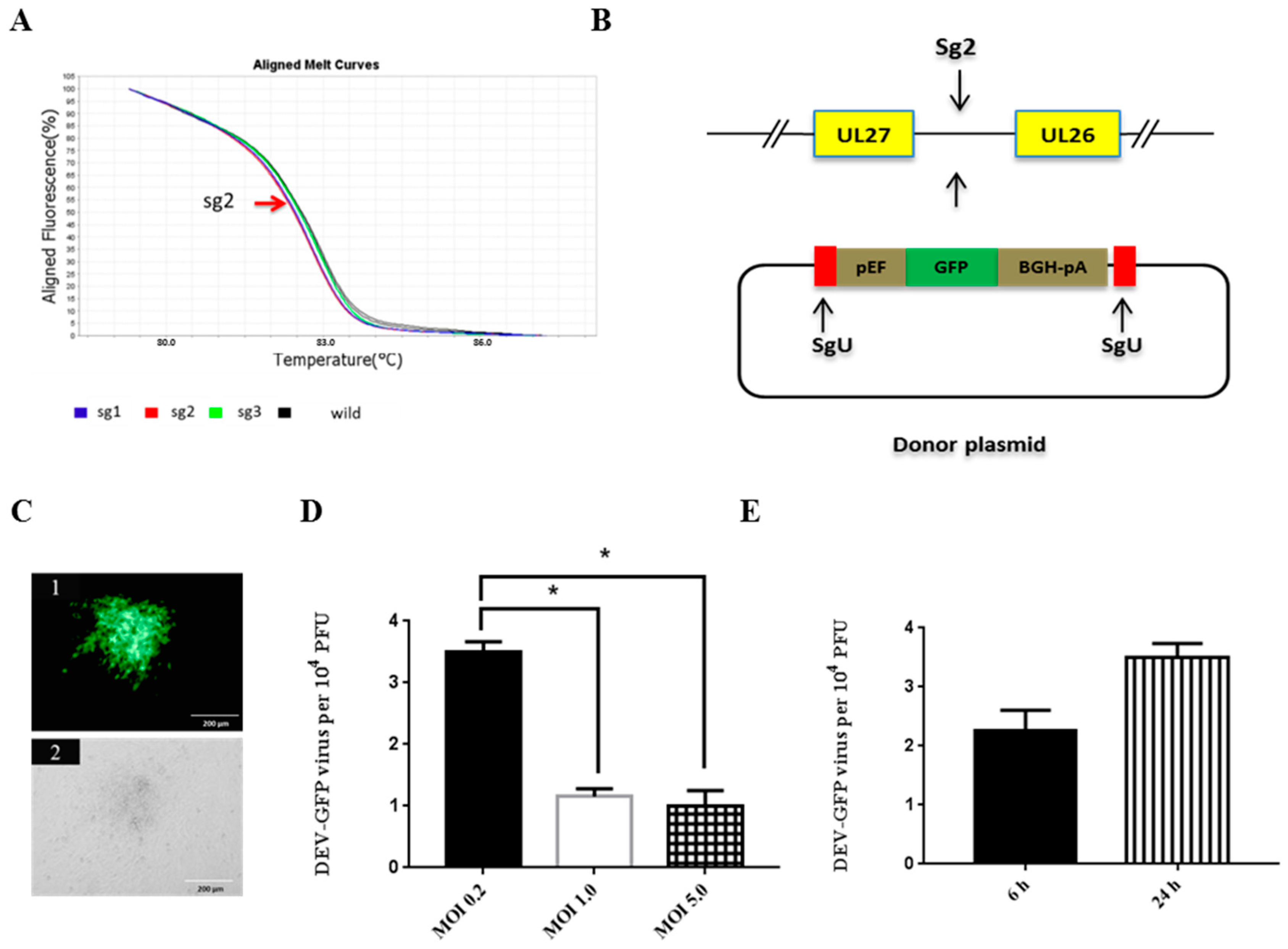
3.1. Optimization of NHEJ-CRISPR/Cas9 System for Gene Knock-In
3.2. NHEJ-CRISPR/Cas9 Based Knock-In Is Non-Directional and the Unintended Indel Is Minimal
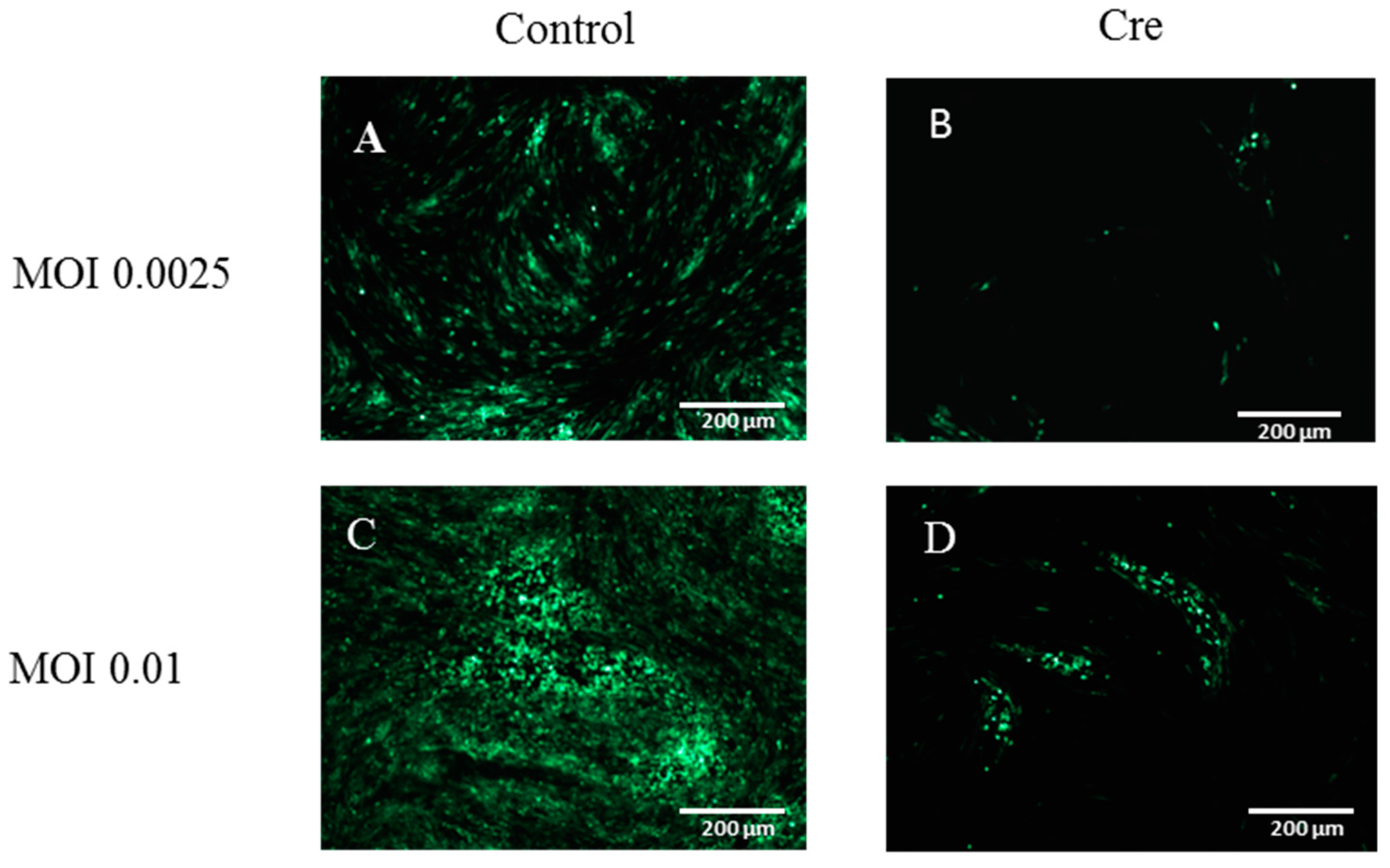
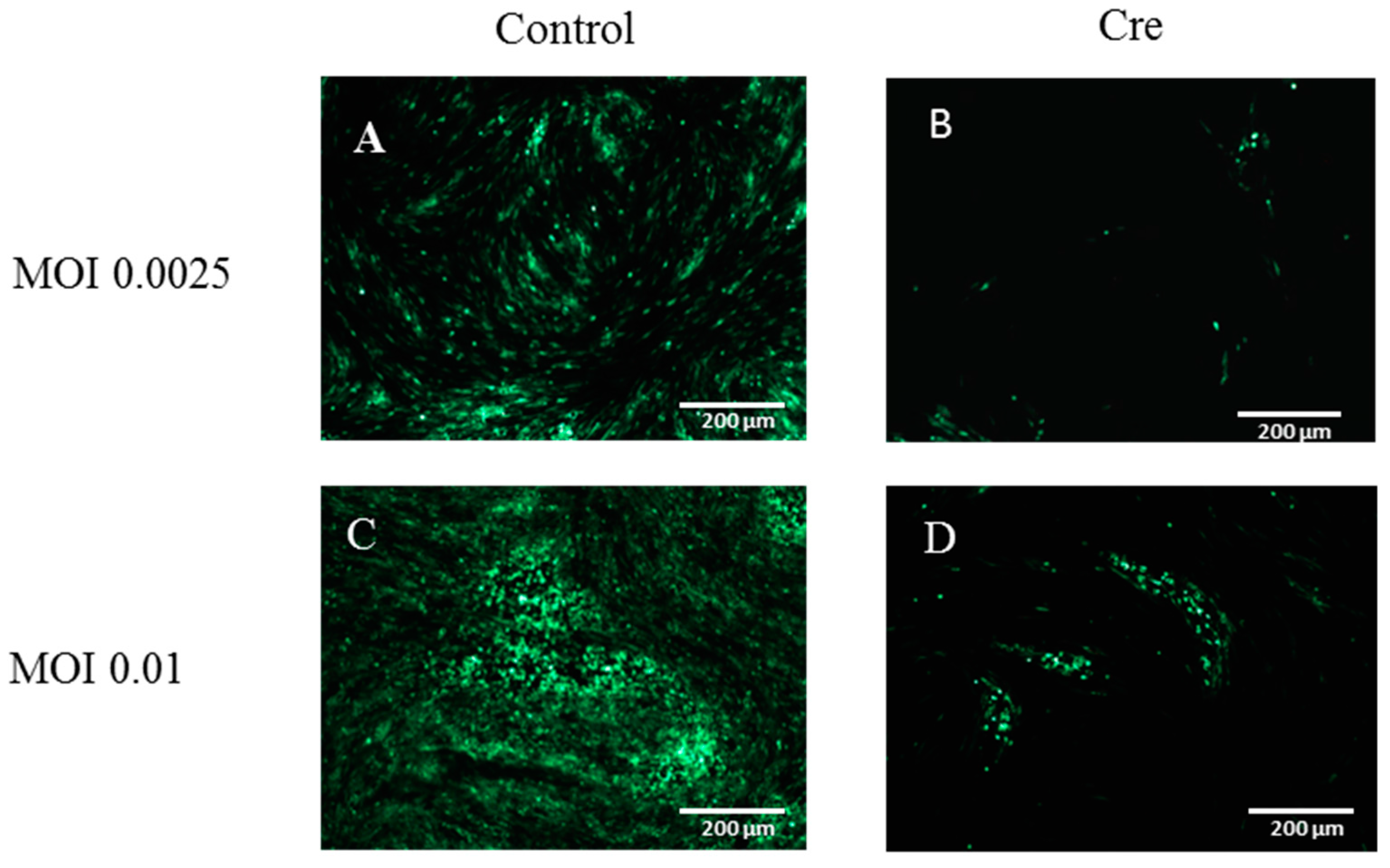
3.3. The Excision of the GFP Cassette from DEV-GFP-HA Is Virus Dose Dependent
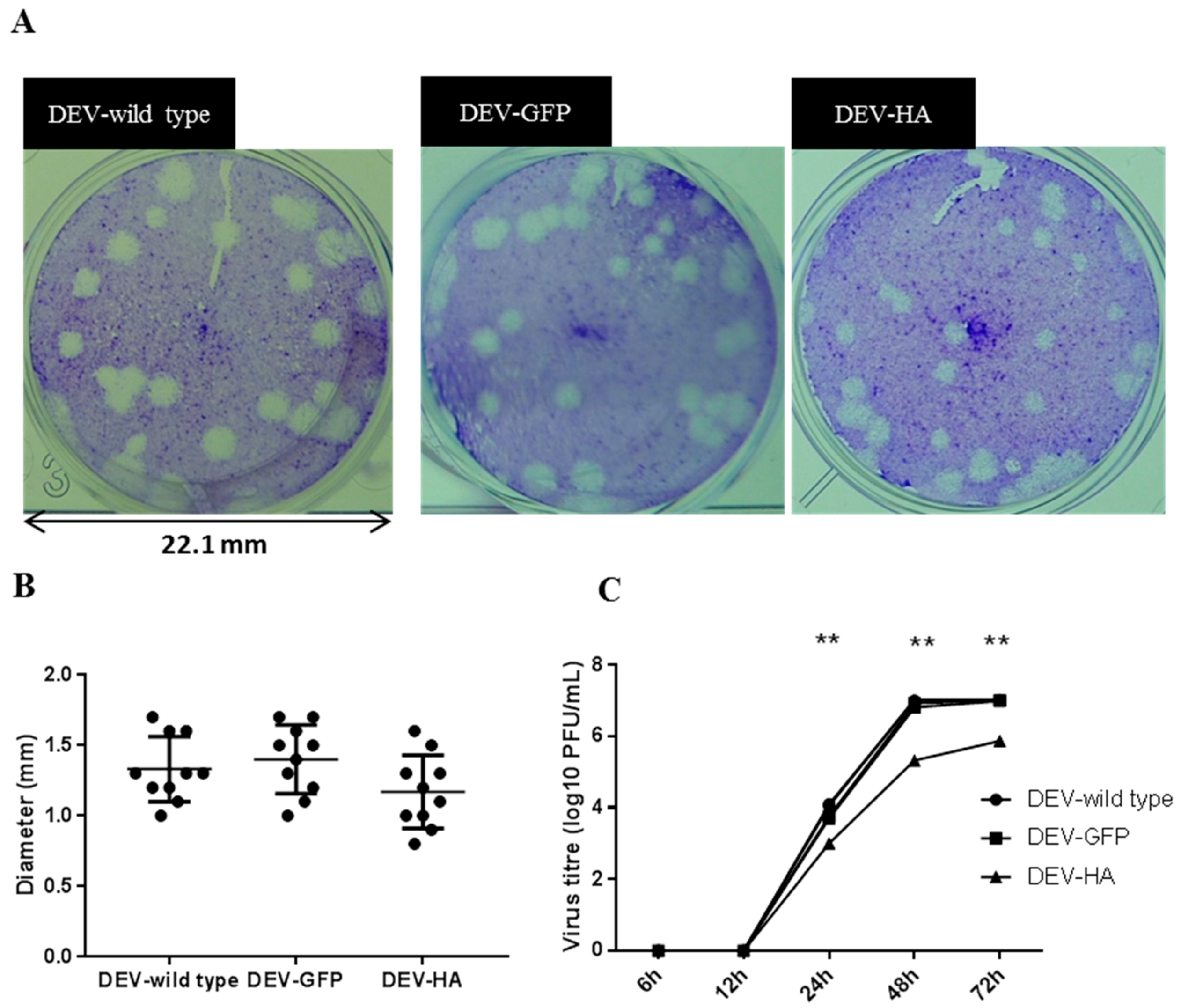
3.4. Characterization of the Recombinant DEV-HA
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dhama, K.; Kumar, N.; Saminathan, M.; Tiwari, R.; Karthik, K.; Kumar, M.A.; Palanivelu, M.; Shabbir, M.Z.; Malik, Y.S.; Singh, R.K. Duck virus enteritis (duck plague)—A comprehensive update. Vet. Q. 2017, 37, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, P.; Jiang, Y.; Wu, L.; Zeng, X.; Tian, G.; Ge, J.; Kawaoka, Y.; Bu, Z.; Chen, H. A duck enteritis virus-vectored bivalent live vaccine provides fast and complete protection against H5N1 avian influenza virus infection in ducks. J. Virol. 2011, 85, 10989–10998. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Huang, K.; Wei, Y.; Chen, H.; Liu, Z.; Jin, M. Construction of a highly efficient CRISPR/Cas9-mediated duck enteritis virus-based vaccine against H5N1 avian influenza virus and duck Tembusu virus infection. Sci. Rep. 2017, 7, 1478. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reddy, K.; Reid, S.M.; Cox, W.J.; Brown, I.H.; Britton, P.; Nair, V.; Iqbal, M. Recombinant herpesvirus of turkeys as a vector-based vaccine against highly pathogenic H7N1 avian influenza and Marek’s disease. Vaccine 2011, 29, 8257–8266. [Google Scholar] [CrossRef] [PubMed]
- Causey, D.; Edwards, S.V. Ecology of avian influenza virus in birds. J. Infect. Dis. 2008, 197 (Suppl. 1), S29–S33. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Negovetich, N.J.; Forrest, H.L.; Webster, R.G. Ducks: The “Trojan horses” of H5N1 influenza. Influenza Other Respir. Viruses 2009, 3, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Pantin-Jackwood, M.J.; Swayne, D.E. Pathobiology of Asian highly pathogenic avian influenza H5N1 virus infections in ducks. Avian Dis. 2007, 51 (Suppl. 1), 250–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ge, A.; Xu, M.; Wang, Z.; Qiao, Y.; Gu, Y.; Liu, C.; Liu, Y.; Hou, J. Construction of a recombinant duck enteritis virus (DEV) expressing hemagglutinin of H5N1 avian influenza virus based on an infectious clone of DEV vaccine strain and evaluation of its efficacy in ducks and chickens. Virol. J. 2015, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, S.; Liu, Y.; Fu, P.; Gao, M.; Mu, X.; Liu, H.; Xing, M.; Ma, B.; Wang, J. Recombinant duck enteritis virus expressing the HA gene from goose H5 subtype avian influenza virus. Vaccine 2013, 31, 5953–5959. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Sun, L.; Yu, T.; Pan, Y.; Wang, D.; Hu, X.; Fu, Z.; He, Q.; Cao, G. A CRISPR/Cas9 and Cre/Lox system-based express vaccine development strategy against re-emerging Pseudorabies virus. Sci. Rep. 2016, 6, 19176. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Hu, Y.; Liu, Z.; Zhong, W.; Cao, H.; Chen, H.; Jin, M. Efficient strategy for constructing duck enteritis virus-based live attenuated vaccine against homologous and heterologous H5N1 avian influenza virus and duck enteritis virus infection. Vet. Res. 2015, 46, 42. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, N.; Hamilton, D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol. 1981, 150, 467–486. [Google Scholar] [CrossRef]
- He, X.; Tan, C.; Wang, F.; Wang, Y.; Zhou, R.; Cui, D.; You, W.; Zhao, H.; Ren, J.; Feng, B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016, 44, e85. [Google Scholar] [CrossRef] [PubMed]
- Dahlem, T.J.; Hoshijima, K.; Jurynec, M.J.; Gunther, D.; Starker, C.G.; Locke, A.S.; Weis, A.M.; Voytas, D.F.; Grunwald, D.J. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012, 8, e1002861. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Qin, C.; Lang, Y.; Wang, M.; Lin, M.; Li, C.; Zhang, R.; Tang, J. A simple and rapid approach to manipulate pseudorabies virus genome by CRISPR/Cas9 system. Biotechnol. Lett. 2015, 37, 1265–1272. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| sgRNA | Target Sequence 5′–3′ | PAM | Gene Locus |
|---|---|---|---|
| Sg1 | GGGTCCAATAACGACCGTCG | TGG | UL26-UL27 |
| Sg2 | GAGCGTATAGTTTAATCGGG | AGG | UL26-UL27 |
| Sg3 | TTTTCCACGACGGTCGTTAT | TGG | UL26-UL27 |
| SgU | GAGATCGAGTGCCGCATCAC | CGG | copGFP |
| Primer Name | Sequence 5′–3′ |
|---|---|
| DEV-HRM-F | TAAAAATTATCCCAAAGCTGTTGCG |
| DEV-HRM-R | CTGGCAAATATGACAACTTTAGCAA |
| DEV-UL26 and 27-F | GGACTTATGCTTTGTATCAAT |
| DEV-UL26 and 27-R | GGGACTAAATTGTTAATTGTTAC |
| Sense-Right-F | GGGAGGATTGGGAAGACAATAG |
| Sense-Right-R | TCCAGAATGTTCAAACGGAGAT |
| Sense-Left-F | GCTGTTGCGTCTCATTGTTG |
| Sense-Left-R | AAGGGCCATAACCCGTAAAG |
| Anti-sense-Right-F | AGCCAATTCCCACTCCTTTC |
| Anti-sense-Left-R | CATCGCATTGTCTGAGTAGGT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, P.; Yao, Y.; Tang, N.; Sadeyen, J.-R.; Sealy, J.; Clements, A.; Bhat, S.; Munir, M.; Bryant, J.E.; Iqbal, M. The Application of NHEJ-CRISPR/Cas9 and Cre-Lox System in the Generation of Bivalent Duck Enteritis Virus Vaccine against Avian Influenza Virus. Viruses 2018, 10, 81. https://doi.org/10.3390/v10020081
Chang P, Yao Y, Tang N, Sadeyen J-R, Sealy J, Clements A, Bhat S, Munir M, Bryant JE, Iqbal M. The Application of NHEJ-CRISPR/Cas9 and Cre-Lox System in the Generation of Bivalent Duck Enteritis Virus Vaccine against Avian Influenza Virus. Viruses. 2018; 10(2):81. https://doi.org/10.3390/v10020081
Chicago/Turabian StyleChang, Pengxiang, Yongxiu Yao, Na Tang, Jean-Remy Sadeyen, Joshua Sealy, Anabel Clements, Sushant Bhat, Muhammad Munir, Juliet E. Bryant, and Munir Iqbal. 2018. "The Application of NHEJ-CRISPR/Cas9 and Cre-Lox System in the Generation of Bivalent Duck Enteritis Virus Vaccine against Avian Influenza Virus" Viruses 10, no. 2: 81. https://doi.org/10.3390/v10020081
APA StyleChang, P., Yao, Y., Tang, N., Sadeyen, J.-R., Sealy, J., Clements, A., Bhat, S., Munir, M., Bryant, J. E., & Iqbal, M. (2018). The Application of NHEJ-CRISPR/Cas9 and Cre-Lox System in the Generation of Bivalent Duck Enteritis Virus Vaccine against Avian Influenza Virus. Viruses, 10(2), 81. https://doi.org/10.3390/v10020081