Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. Primers and Probes
2.3. Construction of Plasmids Carrying Viral Targets
2.4. Multiplex qPCR Standardization
2.5. Patients and Clinical Samples
2.6. Viral Detection in Clinical Samples
2.7. Sanger Sequencing
2.8. Statistical Analysis
3. Results
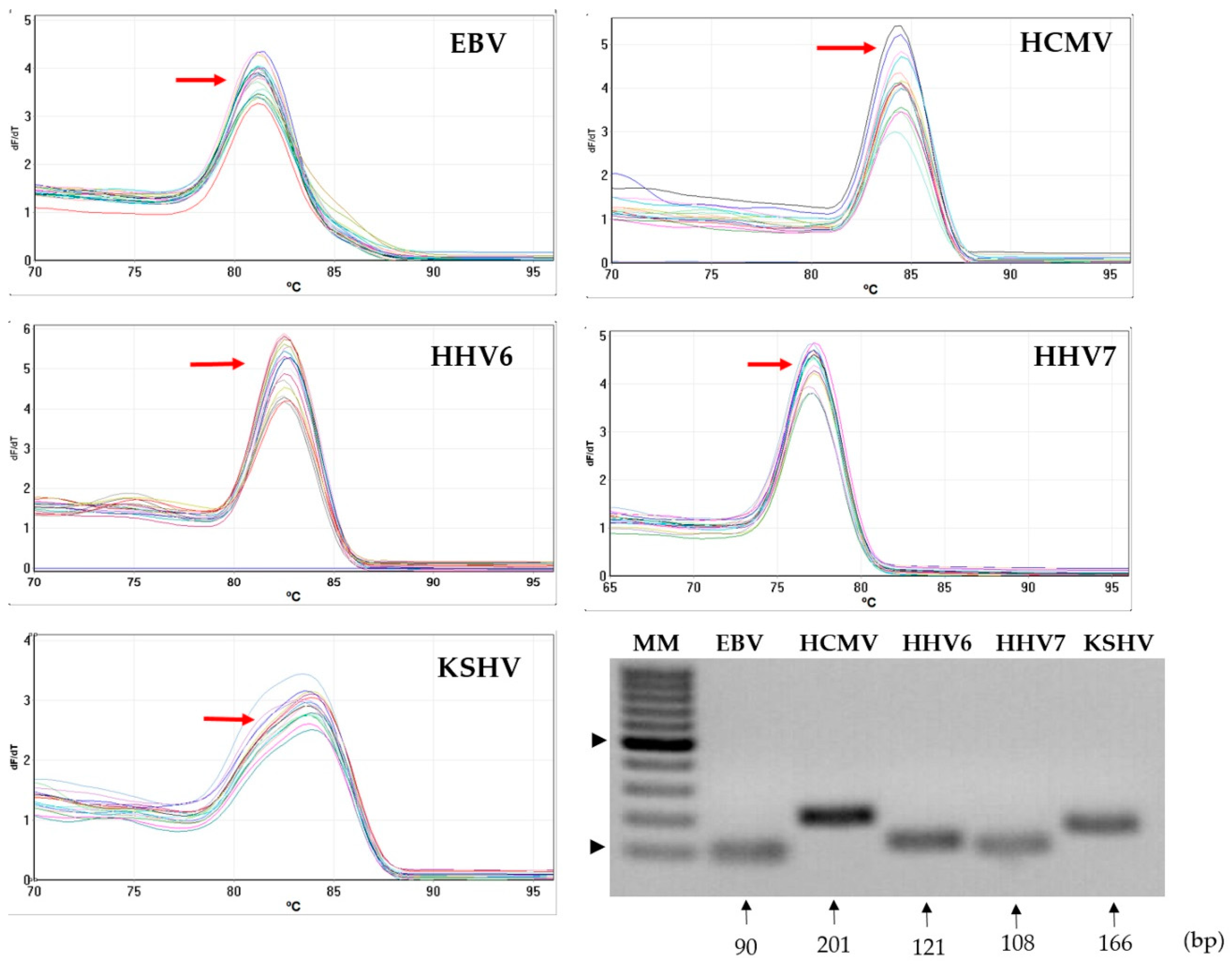
3.1. Specificity of the Primers on a Human DNA Background
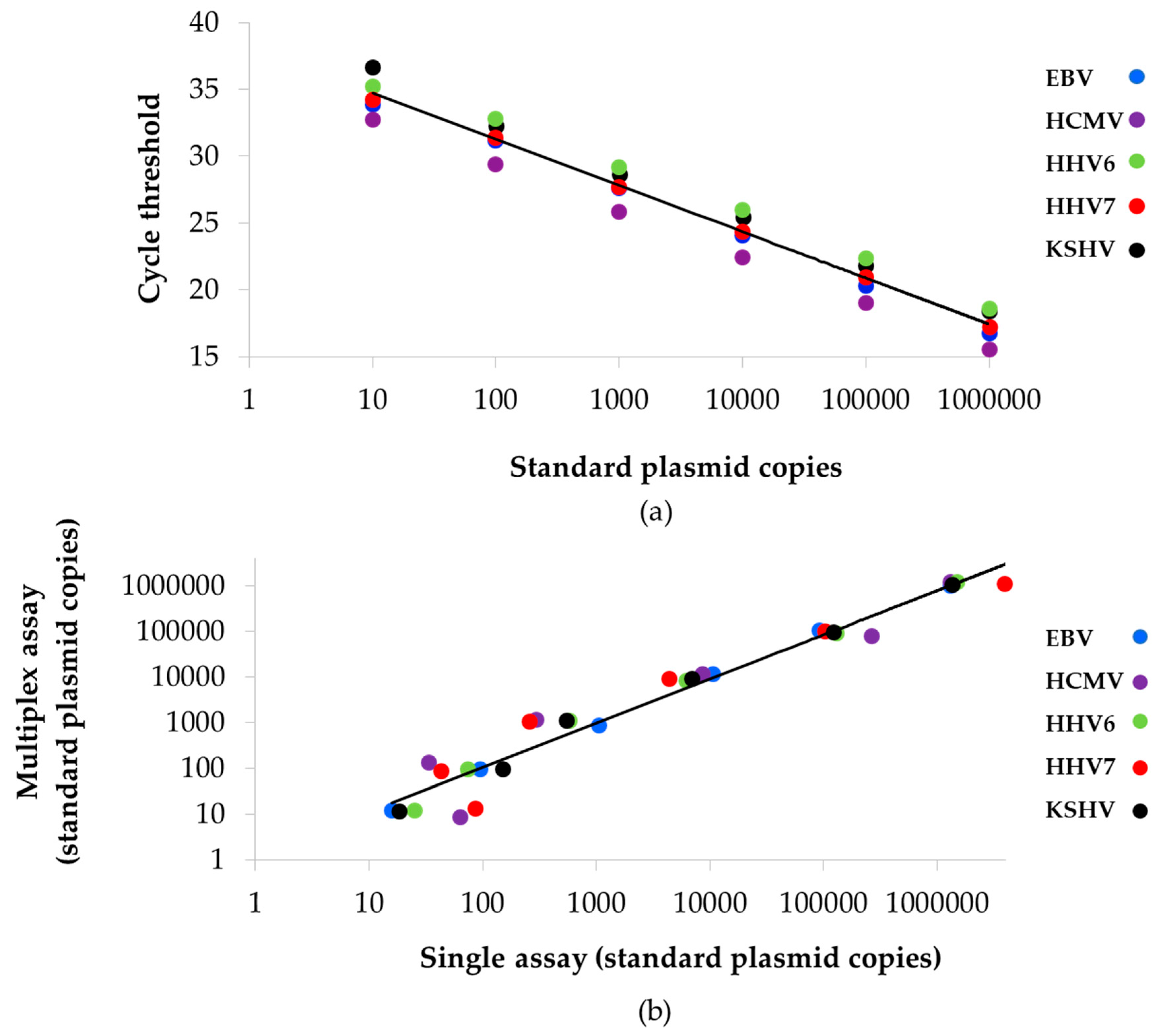
3.2. Performance of the Multiplex qPCRs
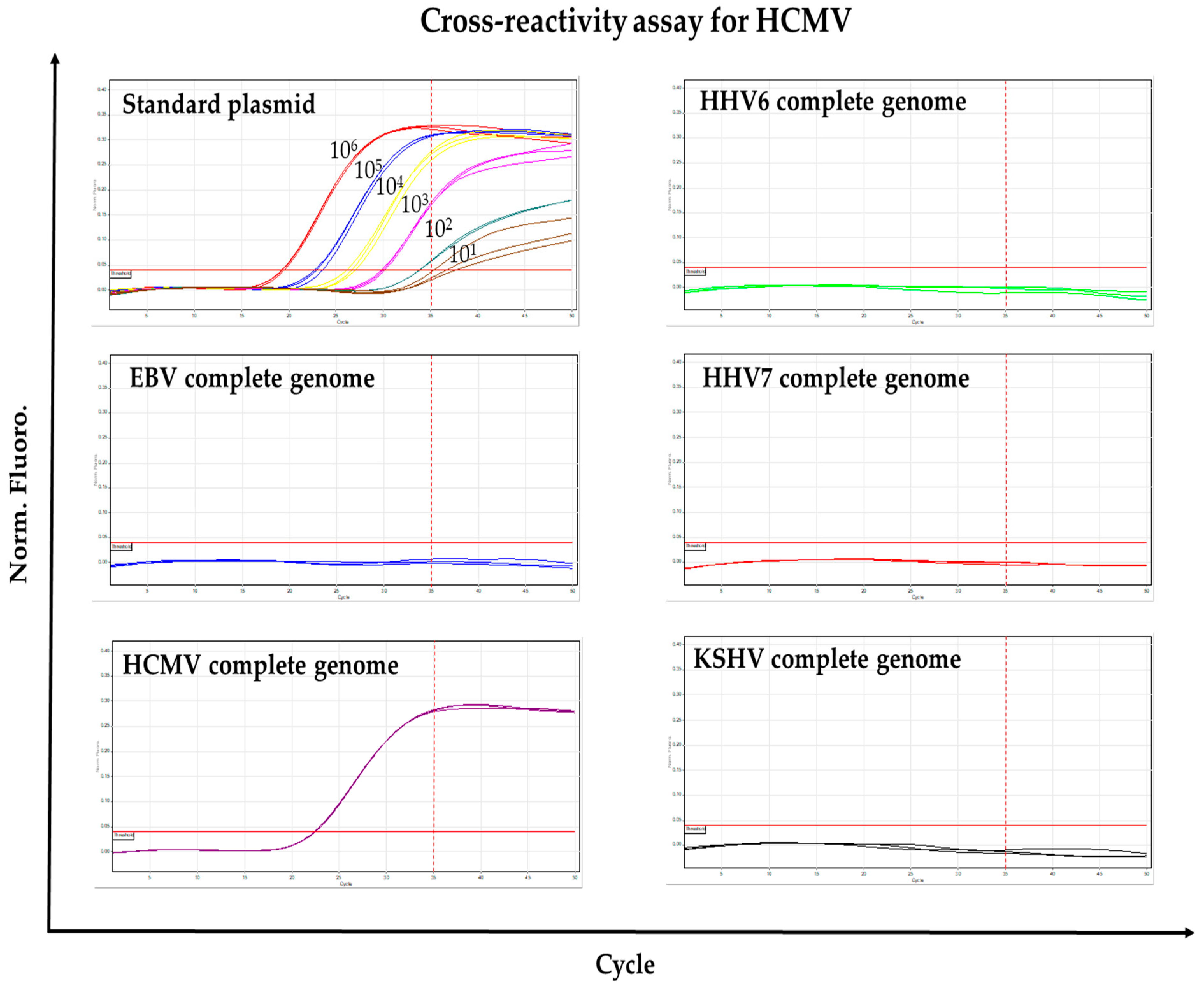
3.3. Cross-Reactivity Test
3.4. Detection of Beta and Gamma Herpesviruses in Pediatric Transplanted Patients
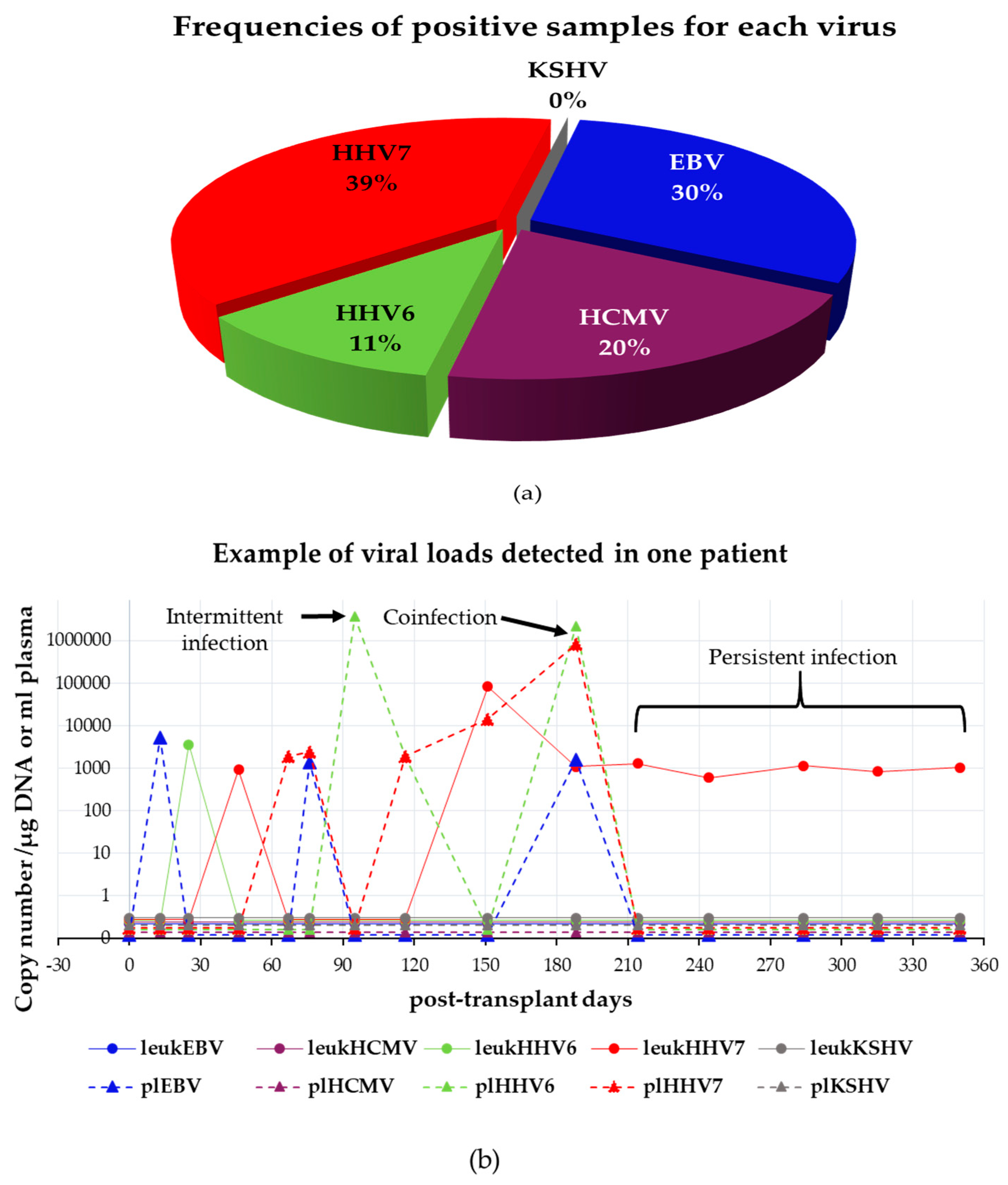
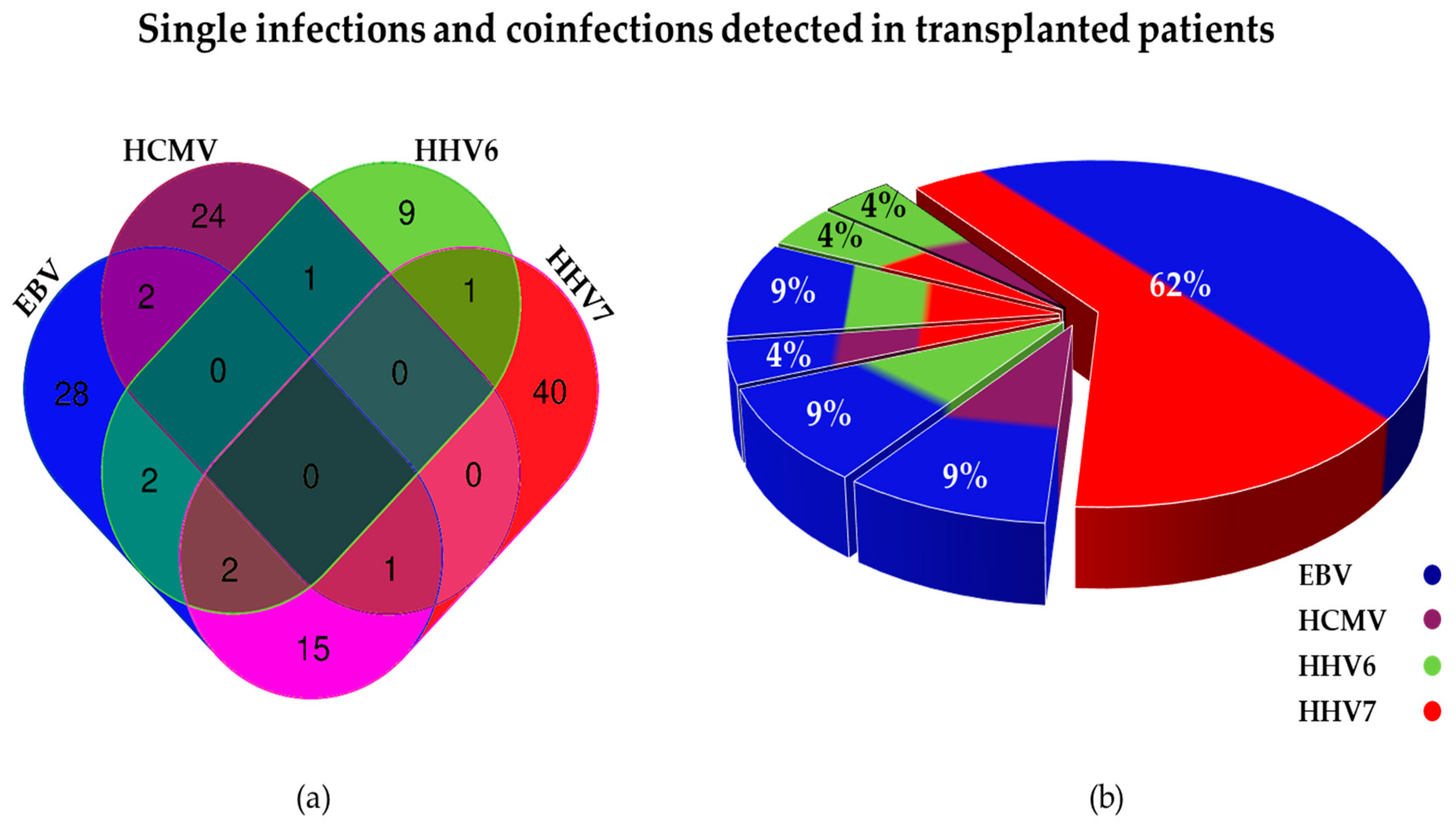
3.4.1. Analysis of Single and Multiple Infections
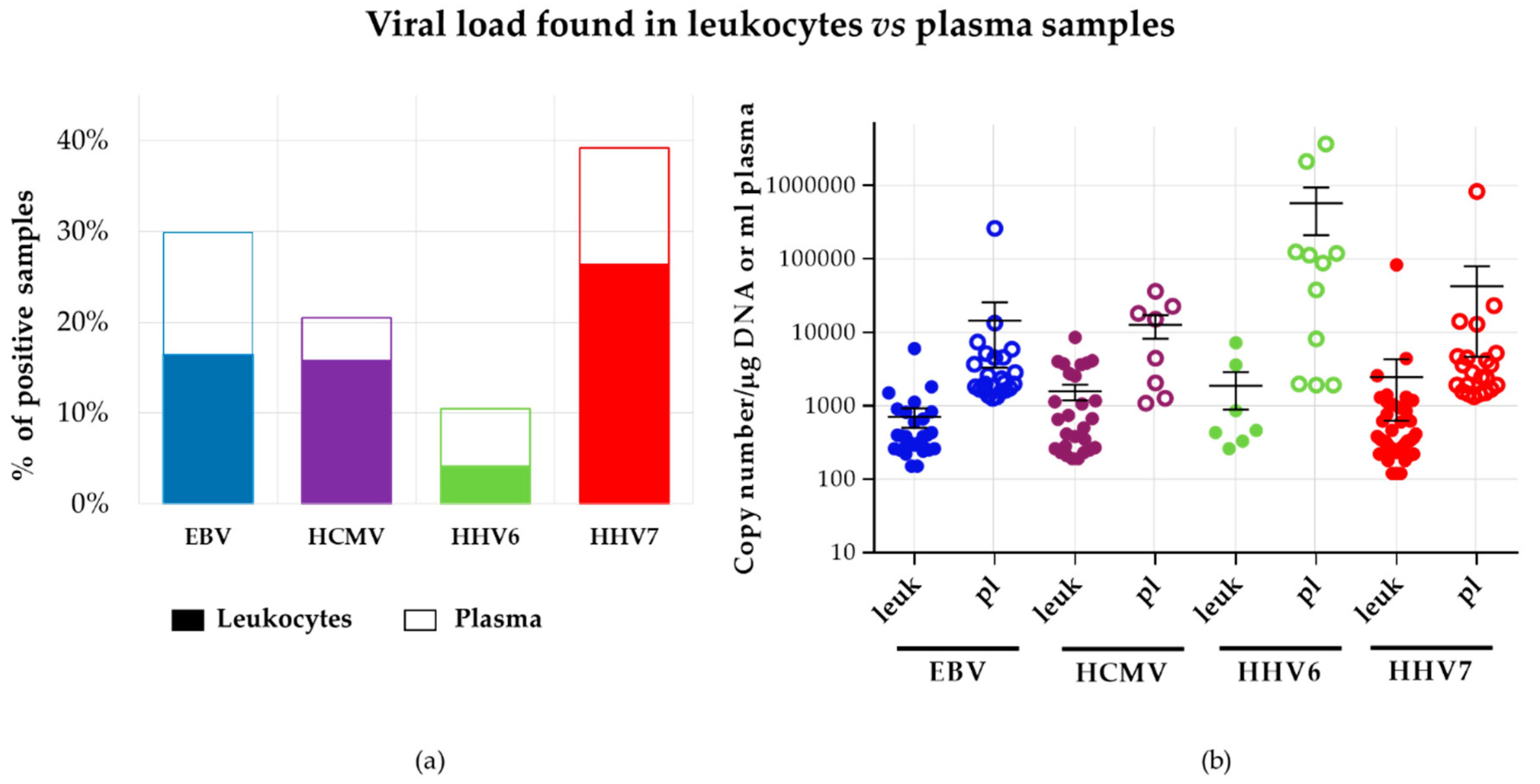
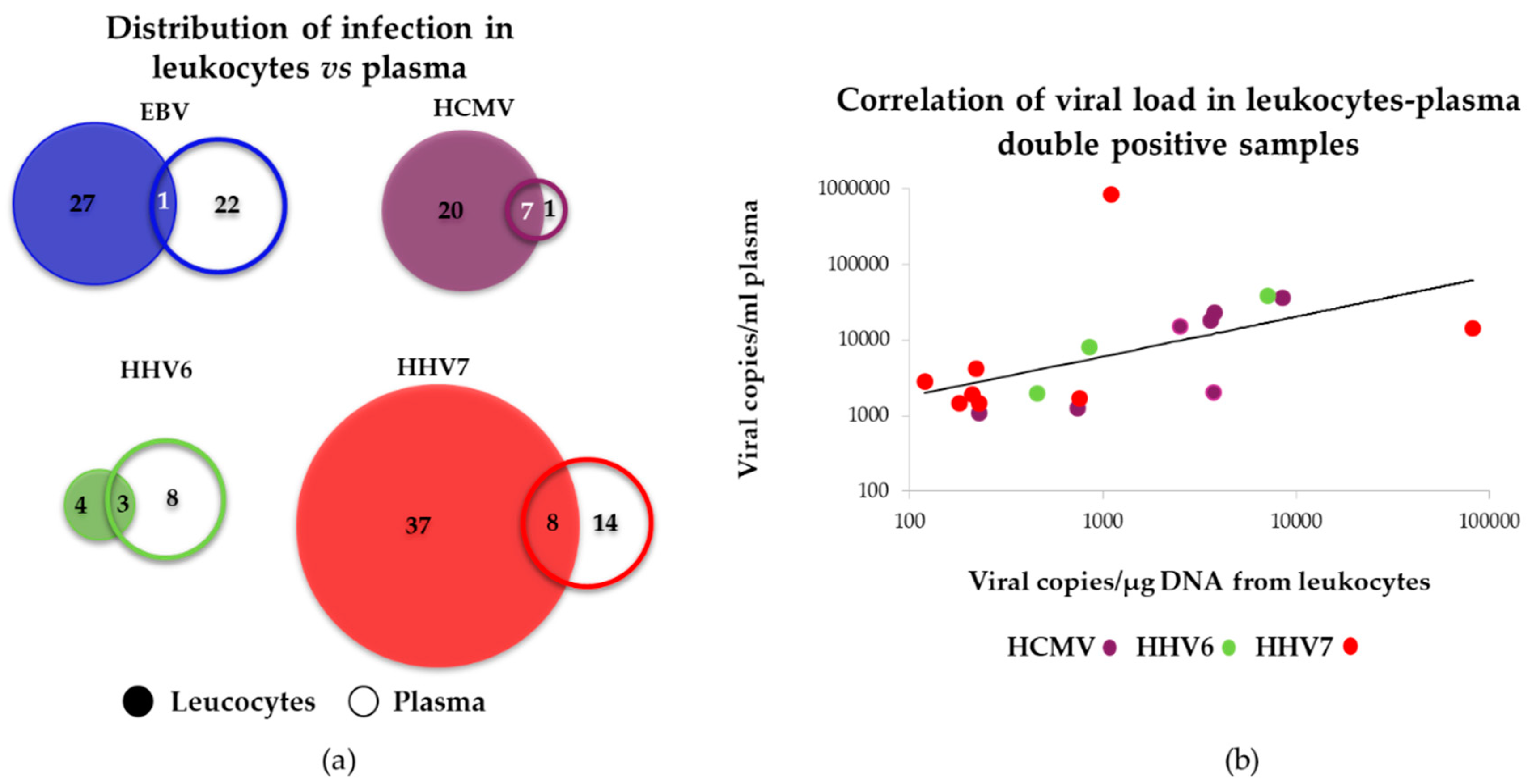
3.4.2. Comparison between Viral Loads Detected in Leukocytes vs. Plasma
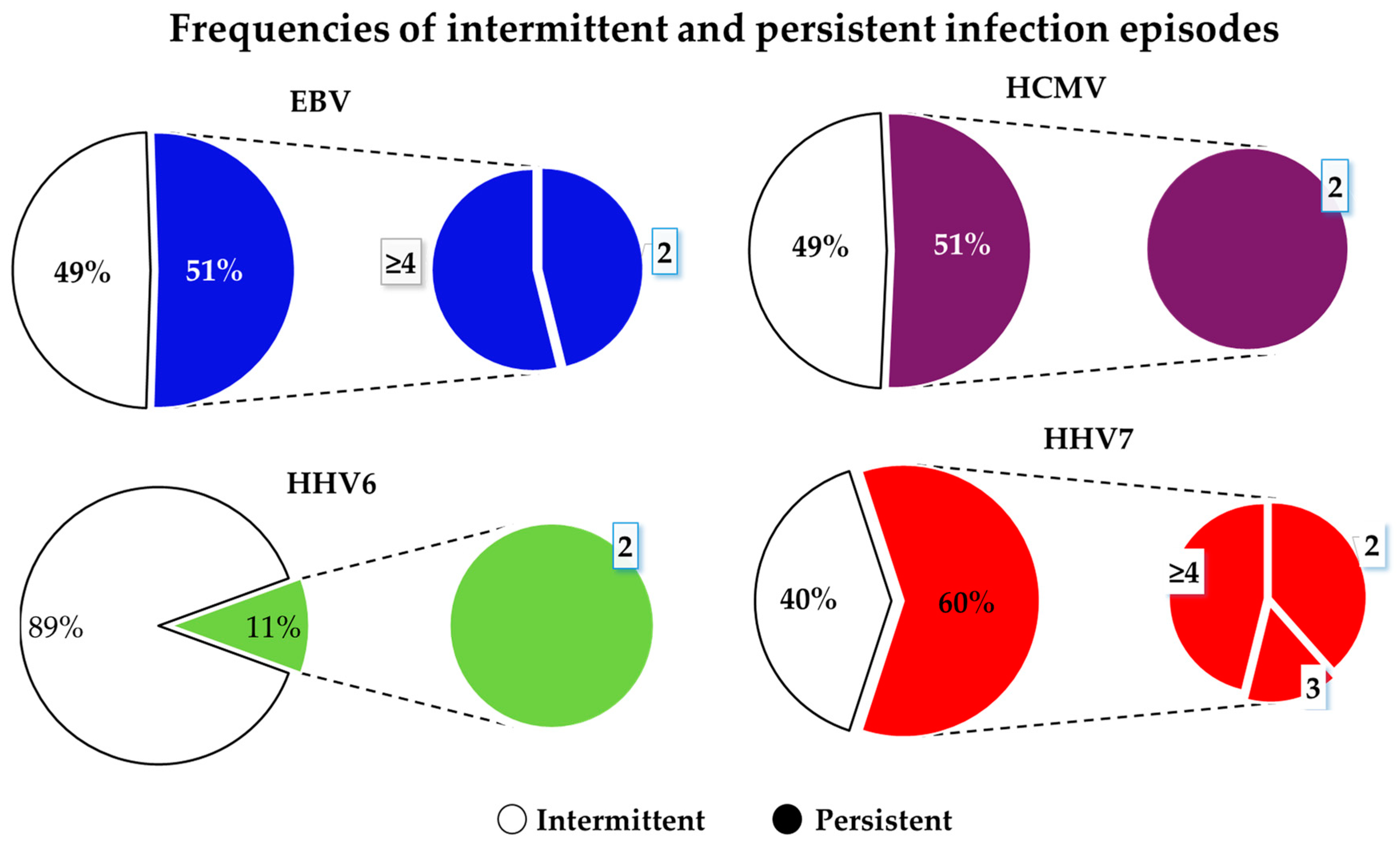
3.4.3. Persistence of Infections
3.4.4. Association between Viral Infection and Episodes of Graft Rejection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Fishman, J.A. Infection in organ transplantation. Am. J. Transplant. 2017, 17, 856–879. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, M.G.; Nador, R.G.; Chadburn, A.; Hyjek, E.M.; Inghirami, G.; Knowles, D.M.; Cesarman, E. Epstein-barr virus latent gene expression in primary effusion lymphomas containing Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8. Blood 1997, 90, 1186–1191. [Google Scholar] [PubMed]
- Vieira, J.; O’Hearn, P.; Kimball, L.; Chandran, B.; Corey, L. Activation of kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 2001, 75, 1378–1386. [Google Scholar] [CrossRef]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct viral and mutational spectrum of endemic burkitt lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Mondragon, M.G.; Carreon-Talavera, R.; Camorlinga-Ponce, M.; Gomez-Delgado, A.; Torres, J.; Fuentes-Panana, E.M. Epstein barr virus and helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS ONE 2013, 8, e62850. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: Future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Wei, C.; Huang, Y.; Zhao, D. Coinfection with EBV/CMV and other respiratory agents in children with suspected infectious mononucleosis. Virol. J. 2010, 7, 247. [Google Scholar] [CrossRef]
- Ono, Y.; Ito, Y.; Kaneko, K.; Shibata-Watanabe, Y.; Tainaka, T.; Sumida, W.; Nakamura, T.; Kamei, H.; Kiuchi, T.; Ando, H.; et al. Simultaneous monitoring by real-time polymerase chain reaction of Epstein-Barr virus, human cytomegalovirus, and human herpesvirus-6 in juvenile and adult liver transplant recipients. Transplant. Proc. 2008, 40, 3578–3582. [Google Scholar] [CrossRef]
- Harma, M.; Hockerstedt, K.; Lyytikainen, O.; Lautenschlager, I. HHV-6 and HHV-7 antigenemia related to CMV infection after liver transplantation. J. Med. Virol. 2006, 78, 800–805. [Google Scholar] [CrossRef]
- Lautenschlager, I.; Lappalainen, M.; Linnavuori, K.; Suni, J.; Höckerstedt, K. CMV infection is usually associated with concurrent HHV-6 and HHV-7 antigenemia in liver transplant patients. J. Clin. Virol. 2002, 25, 57–61. [Google Scholar] [CrossRef]
- Mendez, J.C.; Dockrell, D.H.; Espy, M.J.; Smith, T.F.; Wilson, J.A.; Harmsen, W.S.; Ilstrup, D.; Paya, C.V. Human beta-herpesvirus interactions in solid organ transplant recipients. J. Infect. Dis. 2001, 183, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Humar, A.; Asberg, A.; Kumar, D.; Hartmann, A.; Moussa, G.; Jardine, A.; Rollag, H.; Mouas, H.; Gahlemann, C.G.; Pescovitz, M.D.; et al. An assessment of herpesvirus co-infections in patients with CMV disease: Correlation with clinical and virologic outcomes. Am. J. Transplant. 2009, 9, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, C.T.; Herrmann, M.G.; Gundry, C.N.; Elenitoba-Johnson, K.S. Real-time multiplex PCR assays. Methods 2001, 25, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Bolivar, A.M.; Rojas, A.; Garcia Lugo, P. PCR y pcr-múltiple: Parámetros críticos y protocolo de estandarización. Av. Biomed. 2014, 3, 25–33. [Google Scholar]
- Wada, K.; Kubota, N.; Ito, Y.; Yagasaki, H.; Kato, K.; Yoshikawa, T.; Ono, Y.; Ando, H.; Fujimoto, Y.; Kiuchi, T.; et al. Simultaneous quantification of Epstein-Barr virus, cytomegalovirus, and human herpesvirus 6 DNA in samples from transplant recipients by multiplex real-time PCR assay. J. Clin. Microbiol. 2007, 45, 1426–1432. [Google Scholar] [CrossRef]
- Wada, K.; Mizoguchi, S.; Ito, Y.; Kawada, J.; Yamauchi, Y.; Morishima, T.; Nishiyama, Y.; Kimura, H. Multiplex real-time PCR for the simultaneous detection of herpes simplex virus, human herpesvirus 6, and human herpesvirus 7. Microbiol. Immunol. 2009, 53, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Owczarzy, R.; Tataurov, A.V.; Wu, Y.; Manthey, J.A.; McQuisten, K.A.; Almabrazi, H.G.; Pedersen, K.F.; Lin, Y.; Garretson, J.; McEntaggart, N.O.; et al. Idt scitools: A suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 2008, 36, W163–W169. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Morita, M.; Yabuta, Y.; Kuzushima, K.; Kato, K.; Kojima, S.; Matsuyama, T.; Morishima, T. Quantitative analysis of Epstein-Barr virus load by using a real-time PCR assay. J. Clin. Microbiol. 1999, 37, 132–136. [Google Scholar]
- Tanaka, N.; Kimura, H.; Iida, K.; Saito, Y.; Tsuge, I.; Yoshimi, A.; Matsuyama, T.; Morishima, T. Quantitative analysis of cytomegalovirus load using a real-time PCR assay. J. Med. Virol. 2000, 60, 455–462. [Google Scholar] [CrossRef]
- Hara, S.; Kimura, H.; Hoshino, Y.; Tanaka, N.; Nishikawa, K.; Ihira, M.; Yoshikawa, T.; Morishima, T. Detection of herpesvirus DNA in the serum of immunocompetent children. Microbiol. Immunol. 2002, 46, 177–180. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-blast: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Forootan, A.; Sjoback, R.; Bjorkman, J.; Sjogreen, B.; Linz, L.; Kubista, M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol. Detect. Quantif. 2017, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Reed, G.F.; Lynn, F.; Meade, B.D. Use of coefficient of variation in assessing variability of quantitative assays. Clin. Diagn. Lab. Immunol. 2002, 9, 1235–1239. [Google Scholar] [CrossRef] [PubMed]
- Elnifro, E.M.; Ashshi, A.M.; Cooper, R.J.; Klapper, P.E. Multiplex PCR: Optimization and application in diagnostic virology. Clin. Microbiol. Rev. 2000, 13, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Casas, I.; Pozo, F.; Trallero, G.; Echevarria, J.M.; Tenorio, A. Viral diagnosis of neurological infection by RT multiplex PCR: A search for entero- and herpesviruses in a prospective study. J. Med. Virol. 1999, 57, 145–151. [Google Scholar] [CrossRef]
- Casas, I.; Tenorio, A.; Echevarria, J.M.; Klapper, P.E.; Cleator, G.M. Detection of enteroviral RNA and specific DNA of herpesviruses by multiplex genome amplification. J. Virol. Methods 1997, 66, 39–50. [Google Scholar] [CrossRef]
- Pozo, F.; Tenorio, A. Detection and typing of lymphotropic herpesviruses by multiplex polymerase chain reaction. J. Virol. Methods 1999, 79, 9–19. [Google Scholar] [CrossRef]
- Tanaka, T.; Kogawa, K.; Sasa, H.; Nonoyama, S.; Furuya, K.; Sato, K. Rapid and simultaneous detection of 6 types of human herpes virus (herpes simplex virus, varicella-zoster virus, Epstein-Barr virus, cytomegalovirus, human herpes virus 6a/b, and human herpes virus 7) by multiplex PCR assay. Biomed. Res. 2009, 30, 279–285. [Google Scholar] [CrossRef]
- Tenorio, A.; Echevarria, J.E.; Casas, I.; Echevarria, J.M.; Tabares, E. Detection and typing of human herpesviruses by multiplex polymerase chain reaction. J. Virol. Methods 1993, 44, 261–269. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Stevens, S.J.C.; Verkuijlen, S.A.W.M.; van den Brule, A.J.C.; Middeldorp, J.M. Comparison of quantitative competitive PCR with lightcycler-based PCR for measuring Epstein-Barr virus DNA load in clinical specimens. J. Clin. Microbiol. 2002, 40, 3986–3992. [Google Scholar] [CrossRef] [PubMed]
- Reinke, P.; Prösch, S.; Kern, F.; Volk, H.D. Mechanisms of human cytomegalovirus (HCMV) (re)activation and its impact on organ transplant patients. Transpl. Infect. Dis. 1999, 1, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Marcus, R.; Bradley, J.A. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit. Rev. Oncol. Hematol. 2005, 56, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Almond, P.S.; Matas, A.; Gillingham, K.; Dunn, D.L.; Payne, W.D.; Gores, P.; Gruessner, R.; Najarían, J.S. Risk factor for chronic rejection in renal allograft recipients. Transplantation 1993, 55, 751–757. [Google Scholar] [CrossRef]
- Aumente, M.D. Trasplantes. In Farmacia Hospitalaria; Planas, M.C.G., Ed.; Sociedad Española de Farmacia Hospitalaria: Madrid, Espana, 2002; Volume 26, pp. 1563–1600. [Google Scholar]
- Morales-Sanchez, A.; Fuentes-Panana, E.M. The immunomodulatory capacity of an Epstein-Barr virus abortive lytic cycle: Potential contribution to viral tumorigenesis. Cancers 2018, 10, 98. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tube 1 | Viral/Cellular Target | Gene | F-Q | Sequence 5′→3′ | Amplicon Size (bp) | Ref. |
|---|---|---|---|---|---|---|
| 1 | EBV | BALF5 | FAM-BHQ1a | F CCCTGTTTATCCGATGGAATG R CGGAAGCCCTCTGGACTTC P TGTACACGCACGAGAAATGCGCC | 90 | [19] |
| HCMV | UL123 | JOE-BHQ1a | F GCTACAATAGCCTCTTCCTCATCTG R GACTAGTGTGATGCTGGCCAAG P AGCCTGAGGTTATCAGTGTAATGAAGCGCC | 201 | [20] | |
| Endogenous human gene | β-actin | Cy5-BHQ3a | F CCAGGCTAACCTCGGAAATCT R CATCGTCATTCCTGTGCAACT P TGGGGTGCCGGCTCTCTGCT | 225 | This study | |
| 2 | HHV6 | U31 | FAM-BHQ1a | F CGACTCTCACCCTACTGAACGA R GAGGCTGGCGTCGTAGTAGAA P AGCCACAGCAGCCATCTACATCTGTCAA | 121 | [20] and this study 2 |
| HHV7 | U57 | JOE-BHQ1a | F CGGAAGTCACTGGAGTAATGACAA R ATGCTTTAAACATCCTTTCTTTCGG P CTCGCAGATTGCTTGTTGGCCATG | 108 | [21] | |
| KSHV | LANA | Cy5-BHQ3a | F AGTTATGGGCGACTGGTCTG R GGATGGAAGACGAGATCCAA P AAGTCCGTATGGGTCATTGC | 166 | This study |
| Target | E (%) | Predictability (R2) | LOD (Copy Number) | Correlation of Single vs. Multiplex qPCR (R) |
|---|---|---|---|---|
| EBV | 94 | 0.993 | 21 | 0.9994 |
| HCMV | 92 | 0.988 | 18 | 0.9904 |
| HHV6 | 96 | 0.988 | 25 | 0.9998 |
| HHV7 | 95 | 0.993 | 21 | 0.9981 |
| KSHV | 95 | 0.996 | 18 | 0.9999 |
| Standard (Plasmid Copies) | Coefficients of Variation (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EBV | HCMV | HHV6 | HHV7 | KSHV | ||||||
| Intra 1 | Inter 2 | Intra 1 | Inter 2 | Intra 1 | Inter 2 | Intra 1 | Inter 2 | Intra 1 | Inter 2 | |
| 106 | 0.31 | 0.96 | 0.73 | 3.12 | 0.44 | 1.89 | 0.54 | 0.49 | 0.31 | 0.66 |
| 105 | 0.77 | 1.77 | 2.93 | 2.89 | 0.91 | 1.21 | 0.44 | 0.76 | 0.31 | 0.96 |
| 104 | 0.96 | 1.49 | 1.64 | 2.94 | 0.93 | 1.33 | 0.69 | 0.91 | 1.27 | 1.13 |
| 103 | 1.43 | 1.75 | 3.30 | 2.35 | 0.50 | 1.95 | 1.05 | 0.85 | 0.67 | 0.57 |
| 102 | 1.82 | 1.88 | 1.65 | 2.53 | 0.43 | 1.63 | 0.87 | 0.69 | 0.69 | 1.09 |
| 101 | 2.54 | 2.09 | 3.55 | 3.87 | 0.65 | 0.72 | 1.43 | 2.67 | 1.19 | 2.60 |
| Transplanted Organ | Kidney N = 22 | Liver N = 12 |
|---|---|---|
| Age at transplant | ||
| Median of years (range) | 15 (6–17) | 6 (4–17) |
| Gender | ||
| Female | 33% | 58% |
| Male | 67% | 42% |
| Kind of donor | ||
| Cadaveric | 57% | 92% |
| Living-donor | 43% | 8% |
| EBV serostatus N (%) 1 | ||
| Donor+ Recipient+ | 11 (92%) | 6 (55%) |
| Donor+ Recipient− | 1 (8%) | 2 (18%) |
| Donor− Recipient+ | 0 (0%) | 3 (27%) |
| Donor− Recipient− | 0 (0%) | 0 (0%) |
| HCMV serostatus N (%) 1 | ||
| Donor+ Recipient+ | 13 (62%) | 6 (50%) |
| Donor+ Recipient− | 3 (19%) | 3 (25%) |
| Donor− Recipient+ | 2 (13%) | 2 (17%) |
| Donor− Recipient− | 1 (6%) | 1 (8%) |
| Pre-transplant diagnosis N (%) | ESRD of unknown etiology 15 (68%) Focal and segmental glomerulosclerosis 2 (9%) ESRD secondary to complex uropathy 2 (9%) ESRD secondary to hypoplasia JRA 1 (5%) Microscopic polyangiitis 1 (5%) Posterior urethral valves 1 (5%) | Biliary atresia 3 (25%) |
| Fulminant hepatitis 2 (17%) | ||
| Liver fibrosis 1 (8%) | ||
| Neonatal giant cell hepatitis 1 (8%) | ||
| Progressive intrahepatic family cholestasis 1 (8%) | ||
| Bayler disease 1 (8%) | ||
| Alalgille syndrome 1 (8%) | ||
| Tyrosinemia 1 (8%) | ||
| Cholesterol ester deposition 1 (8%) |
| Infection/Coinfection | Kidney | Liver | ||
|---|---|---|---|---|
| Relative Risk | p Value | Relative Risk | p Value | |
| EBV | 2.74 | 0.20 | 2.68 | 0.41 |
| HCMV | 4.28 | 0.06 | 40.33 | 0.0013 |
| HHV6 | 5.60 | 0.03 | 6 | 0.20 |
| HHV7 | 3.04 | 0.12 | 1.27 | 0.87 |
| EBV/HCMV | 4.86 | 0.24 | NOT DETECTED | |
| EBV/HHV6 | 4.86 | 0.24 | 9.07 | 0.10 |
| EBV/HHV7 | 5.60 | 0.03 | 4.46 | 0.29 |
| HCMV/HHV6 | 7.32 | 0.12 | NOT DETECTED | |
| HCMV/HHV7 | NOT DETECTED | NOT DETECTED | ||
| HHV6/HHV7 | 4.86 | 0.24 | NOT DETECTED | |
| EVB/HCMV/HHV6 | NOT DETECTED | NOT DETECTED | ||
| EBV/HCMV/HHV7 | 7.32 | 0.12 | NOT DETECTED | |
| EBV/HHV6/HHV7 | 17.64 | 0.0003 | NOT DETECTED | |
| HCMV/HHV6/HHV7 | NOT DETECTED | NOT DETECTED | ||
| EBV/HCMV/HHV6/HHV7 | NOT DETECTED | NOT DETECTED | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Ponce, Y.; Varela-Fascinetto, G.; Romo-Vázquez, J.C.; López-Martínez, B.; Sánchez-Huerta, J.L.; Parra-Ortega, I.; Fuentes-Pananá, E.M.; Morales-Sánchez, A. Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation. Viruses 2018, 10, 730. https://doi.org/10.3390/v10120730
Sánchez-Ponce Y, Varela-Fascinetto G, Romo-Vázquez JC, López-Martínez B, Sánchez-Huerta JL, Parra-Ortega I, Fuentes-Pananá EM, Morales-Sánchez A. Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation. Viruses. 2018; 10(12):730. https://doi.org/10.3390/v10120730
Chicago/Turabian StyleSánchez-Ponce, Yessica, Gustavo Varela-Fascinetto, José Carlos Romo-Vázquez, Briceida López-Martínez, José Luis Sánchez-Huerta, Israel Parra-Ortega, Ezequiel M. Fuentes-Pananá, and Abigail Morales-Sánchez. 2018. "Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation" Viruses 10, no. 12: 730. https://doi.org/10.3390/v10120730
APA StyleSánchez-Ponce, Y., Varela-Fascinetto, G., Romo-Vázquez, J. C., López-Martínez, B., Sánchez-Huerta, J. L., Parra-Ortega, I., Fuentes-Pananá, E. M., & Morales-Sánchez, A. (2018). Simultaneous Detection of Beta and Gamma Human Herpesviruses by Multiplex qPCR Reveals Simple Infection and Coinfection Episodes Increasing Risk for Graft Rejection in Solid Organ Transplantation. Viruses, 10(12), 730. https://doi.org/10.3390/v10120730