EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis


{kind=link}
{kind=link}
Abstract
1. Introduction
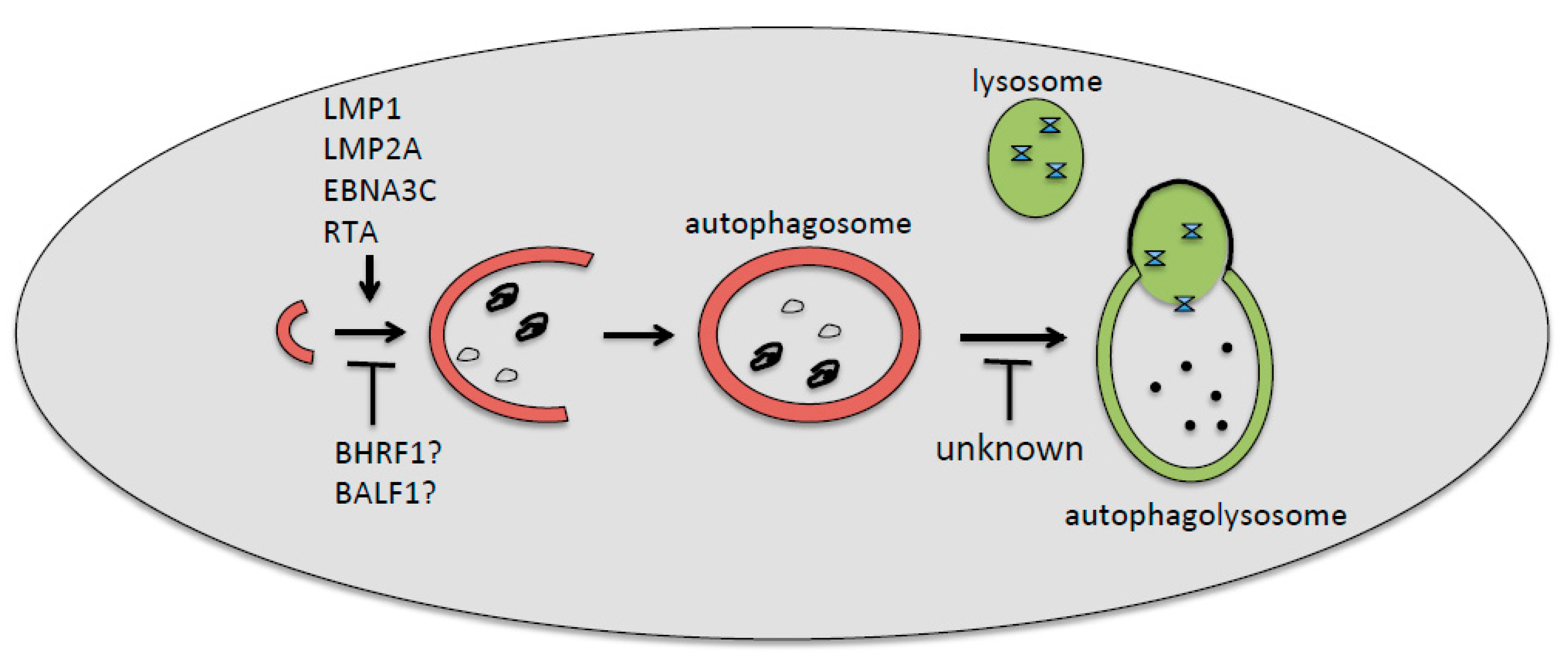
2. EBV De Novo Infection and Autophagy
3. EBV Lytic Reactivation and Autophagy
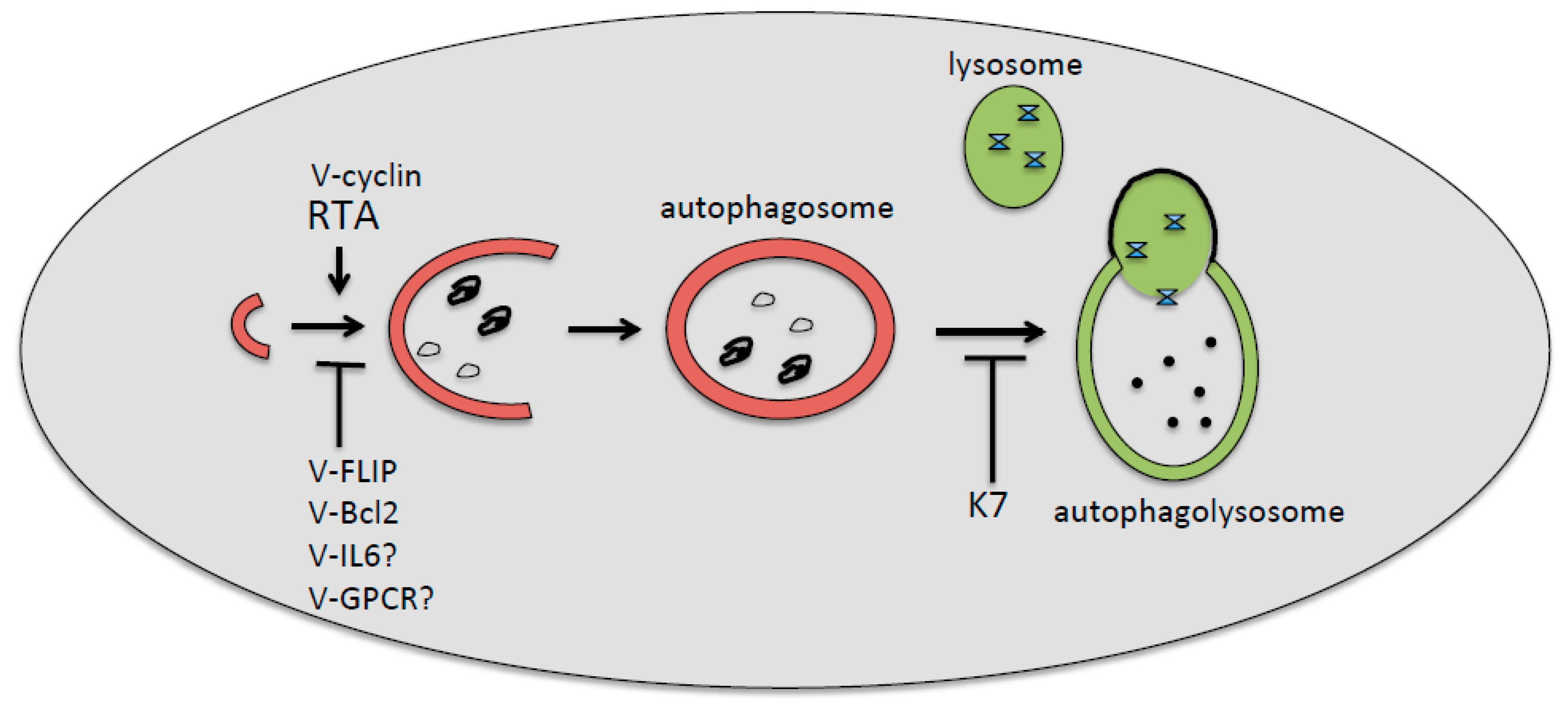
4. KSHV De Novo Infection and Autophagy
5. KSHV Lytic Reactivation and Autophagy
6. Conclusions
Funding
Conflicts of Interest
References
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic epstein-barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; Dabral, P.; Gupta, N.; Sarkar, R.; Verma, S.C. KSHV genome replication and maintenance. Front. Microbiol. 2016, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Joo, E.H.; Song, K.A.; Choi, B.; Kim, M.; Kim, S.H.; Kim, S.J.; Kang, M.S. Effects of lymphocyte profile on development of EBV-induced lymphoma subtypes in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Dissinger, N.J.; Damania, B. Recent advances in understanding Kaposi’s sarcoma-associated herpesvirus. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; Heston, L.; Grogan, E.; Gradoville, L.; Rigsby, M.; Sun, R.; Shedd, D.; Kushnaryov, V.M.; Grossberg, S.; Chang, Y. Selective switch between latency and lytic replication of Kaposi’s sarcoma herpesvirus and epstein-barr virus in dually infected body cavity lymphoma cells. J. Virol. 1997, 71, 314–324. [Google Scholar] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Filardi, M.; Gonnella, R.; Farina, A.; Torrisi, M.R.; Faggioni, A.; Cirone, M. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy 2015, 11, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Autophagy and disease. J. Biol. Chem. 2018, 293, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Stepping back from the guidelines: Where do we stand? Autophagy 2016, 12, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, L.; Jones, C.A.; Abendroth, A.; Cunningham, A.L. Dendritic cell biology in herpesvirus infections. Viral Immunol. 2005, 18, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 2009, 83, 12164–12171. [Google Scholar] [CrossRef] [PubMed]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 2011, 85, 9414–9424. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.; Peruzzi, G.; Lacconi, V.; Lenna, S.; Quarta, S.; Rosato, E.; Vestri, A.R.; York, M.; Dreyfus, D.H.; Faggioni, A.; et al. Epstein-Barr virus lytic infection promotes activation of toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res. Ther. 2017, 19, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, D.; Hutt-Fletcher, L.; Morgan, A.; Masucci, M.G.; Levitsky, V. Epstein-Barr virus inhibits the development of dendritic cells by promoting apoptosis of their monocyte precursors in the presence of granulocyte macrophage-colony-stimulating factor and interleukin-4. Blood 2002, 99, 3725–3734. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2 induces autophagy to promote abnormal acinus formation. J. Virol. 2015, 89, 6940–6944. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. 2018, 9, 605. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Fathallah, I.; Gruffat, H.; Mariggio, G.; Le Calvez-Kelm, F.; Voegele, C.; Bartosch, B.; Hernandez-Vargas, H.; McKay, J.; Sylla, B.S.; et al. Epstein-barr virus transforming protein LMP-1 alters B cells gene expression by promoting accumulation of the oncoprotein ΔNp73α. PLoS Pathog. 2013, 9, e1003186. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Hafez, A.Y.; Kishton, R.; Messinger, J.E.; Nikitin, P.A.; Rathmell, J.C.; Luftig, M.A. Metabolic stress is a barrier to Epstein-Barr virus-mediated B-cell immortalization. Proc. Natl. Acad. Sci. USA 2016, 113, E782–E790. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.L.; Xu, S.; Lyons-Weiler, J.; Rosendorff, A.; Webber, S.A.; Wasil, L.R.; Metes, D.; Rowe, D.T. Cellular factors associated with latency and spontaneous epstein-barr virus reactivation in B-lymphoblastoid cell lines. Virology 2010, 400, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. BCL-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- E, X.; Hwang, S.; Oh, S.; Lee, J.S.; Jeong, J.H.; Gwack, Y.; Kowalik, T.F.; Sun, R.; Jung, J.U.; Liang, C. Viral BCL-2-mediated evasion of autophagy aids chronic infection of gammaherpesvirus 68. PLoS Pathog. 2009, 5, e1000609. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kroemer, G. Defective autophagy initiates malignant transformation. Mol. Cell 2016, 62, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Antigen processing for MHC class II presentation via autophagy. Front. Immunol. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Severa, M.; Giacomini, E.; Gafa, V.; Anastasiadou, E.; Rizzo, F.; Corazzari, M.; Romagnoli, A.; Trivedi, P.; Fimia, G.M.; Coccia, E.M. EBV stimulates TLR- and autophagy-dependent pathways and impairs maturation in plasmacytoid dendritic cells: Implications for viral immune escape. Eur. J. Immunol. 2013, 43, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Santarelli, R.; Granato, M.; Gonnella, R.; Torrisi, M.R.; Faggioni, A.; Cirone, M. EBV reduces autophagy, intracellular ROS and mitochondria to impair monocyte survival and differentiation. Autophagy 2018. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Zaccagnino, P.; Di Paola, M.; Saltarella, M.; Oliveros Celis, C.; Nico, B.; Santoro, G.; Lorusso, M. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic. Biol. Med. 2008, 44, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Chen, L.W.; Wang, W.H.; Chang, P.J.; Chiu, Y.F.; Hung, C.C.; Lin, Y.J.; Liou, J.Y.; Tsai, W.J.; Hung, C.L.; et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Chiu, Y.F.; Wang, W.H.; Chang, L.K.; Liu, S.T. Activation of the ERK signal transduction pathway by Epstein-Barr virus immediate-early protein Rta. J. Gen. Virol. 2008, 89, 2437–2446. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [PubMed]
- Gonnella, R.; Granato, M.; Farina, A.; Santarelli, R.; Faggioni, A.; Cirone, M. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochim. Biophys. Acta 2015, 1853, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052. [Google Scholar] [CrossRef] [PubMed]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Munz, C. Macroautophagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Flaherty, S.; Veettil, M.V.; Johnson, K.E.; Chandran, B.; Bottero, V. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 activation in latently infected endothelial cells through SQSTM1 phosphorylation and interaction with polyubiquitinated keap1. J. Virol. 2015, 89, 2268–2286. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Lampe, M.; Gunther, T.; Brune, W. The viral Bcl-2 homologs of kaposi’s sarcoma-associated herpesvirus and rhesus rhadinovirus share an essential role for viral replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Granato, M.; Pentassuglia, G.; Lacconi, V.; Gilardini Montani, M.S.; Gonnella, R.; Tafani, M.; Torrisi, M.R.; Faggioni, A.; Cirone, M. KSHV reduces autophagy in THP-1 cells and in differentiating monocytes by decreasing CAST/calpastatin and ATG5 expression. Autophagy 2016, 12, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Wang, H.; Nicholas, J. Human herpesvirus 8 interleukin-6 (viL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J. Virol. 1999, 73, 8268–8278. [Google Scholar] [PubMed]
- Santarelli, R.; Gonnella, R.; Di Giovenale, G.; Cuomo, L.; Capobianchi, A.; Granato, M.; Gentile, G.; Faggioni, A.; Cirone, M. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci. Rep. 2014, 4, 4241. [Google Scholar] [CrossRef] [PubMed]
- Suthaus, J.; Stuhlmann-Laeisz, C.; Tompkins, V.S.; Rosean, T.R.; Klapper, W.; Tosato, G.; Janz, S.; Scheller, J.; Rose-John, S. HHV-8-encoded viral IL-6 collaborates with mouse IL-6 in the development of multicentric castleman disease in mice. Blood 2012, 119, 5173–5181. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bhaduri-McIntosh, S. A central role for STAT3 in gammaherpesvirus-life cycle and -diseases. Front. Microbiol. 2016, 7, 1052. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Gilardini Montani, M.S.; Santarelli, R.; D’Orazi, G.; Faggioni, A.; Cirone, M. Apigenin, by activating p53 and inhibiting STAT3, modulates the balance between pro-apoptotic and pro-survival pathways to induce PEL cell death. J. Exp. Clin. Cancer Res. 2017, 36, 167. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Kepp, O.; Criollo, A.; Vicencio, J.M.; Soussi, T.; Kroemer, G. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008, 7, 3056–3061. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ueda, K.; Guwanan, E.; Sakakibara, S.; Do, E.; Osaki, E.; Yada, K.; Okuno, T.; Yamanishi, K. A posttranscriptional regulator of Kaposi’s sarcoma-associated herpesvirus interacts with RNA-binding protein PCBP1 and controls gene expression through the IRES. Virology 2004, 325, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. P53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi’s sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 2012, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Cheng, A.; Zou, Z.; Yang, Y.S.; Sumpter, R.M., Jr.; Huang, C.L.; Bhagat, G.; Virgin, H.W.; Lira, S.A.; Levine, B. Endolysosomal trafficking of viral G protein-coupled receptor functions in innate immunity and control of viral oncogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, 2994–2999. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.G.; Sadowska, M.; Reid, W.; Tschachler, E.; Hayward, G.; Reitz, M. Kaposi’s sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J. Virol. 2003, 77, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Sin, S.H.; Lucas, A.; Venkataramanan, R.; Wang, L.; Eason, A.; Chavakula, V.; Hilton, I.B.; Tamburro, K.M.; Damania, B.; et al. mTOR inhibitors block Kaposi sarcoma growth by inhibiting essential autocrine growth factors and tumor angiogenesis. Cancer Res. 2013, 73, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dong, K.; Liang, W.; Xu, D.; Xia, H.; Geng, J.; Najafov, A.; Liu, M.; Li, Y.; Han, X.; et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. eLife 2015, 4, e06734. [Google Scholar] [CrossRef] [PubMed]
- Masuelli, L.; Granato, M.; Benvenuto, M.; Mattera, R.; Bernardini, R.; Mattei, M.; d’Amati, G.; D’Orazi, G.; Faggioni, A.; Bei, R.; et al. Chloroquine supplementation increases the cytotoxic effect of curcumin against Her2/neu overexpressing breast cancer cells in vitro and in vivo in nude mice while counteracts it in immune competent mice. Oncoimmunology 2017, 6, e1356151. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirone, M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 2018, 10, 599. https://doi.org/10.3390/v10110599
Cirone M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses. 2018; 10(11):599. https://doi.org/10.3390/v10110599
Chicago/Turabian StyleCirone, Mara. 2018. "EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis" Viruses 10, no. 11: 599. https://doi.org/10.3390/v10110599
APA StyleCirone, M. (2018). EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses, 10(11), 599. https://doi.org/10.3390/v10110599