New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Selection of Clinical DNA Isolates for NGS
2.3. Sequencing
2.3.1. Illumina Short-Read Sequencing
2.3.2. Oxford Nanopore Technologies (ONT) Long-Read Sequencing
2.4. Sequence-Data Processing
2.4.1. Short-Read Data Pre-Processing
2.4.2. Long-Read Data Pre-Processing
2.4.3. Genome Assembly
2.4.4. Genome Annotation
2.5. Diversity Estimation and Phylogenetic Trees
2.6. Evaluation of Genome Mosaicity and Recombination
3. Results and Discussion
3.1. MCV Genome Assembly and Annotation
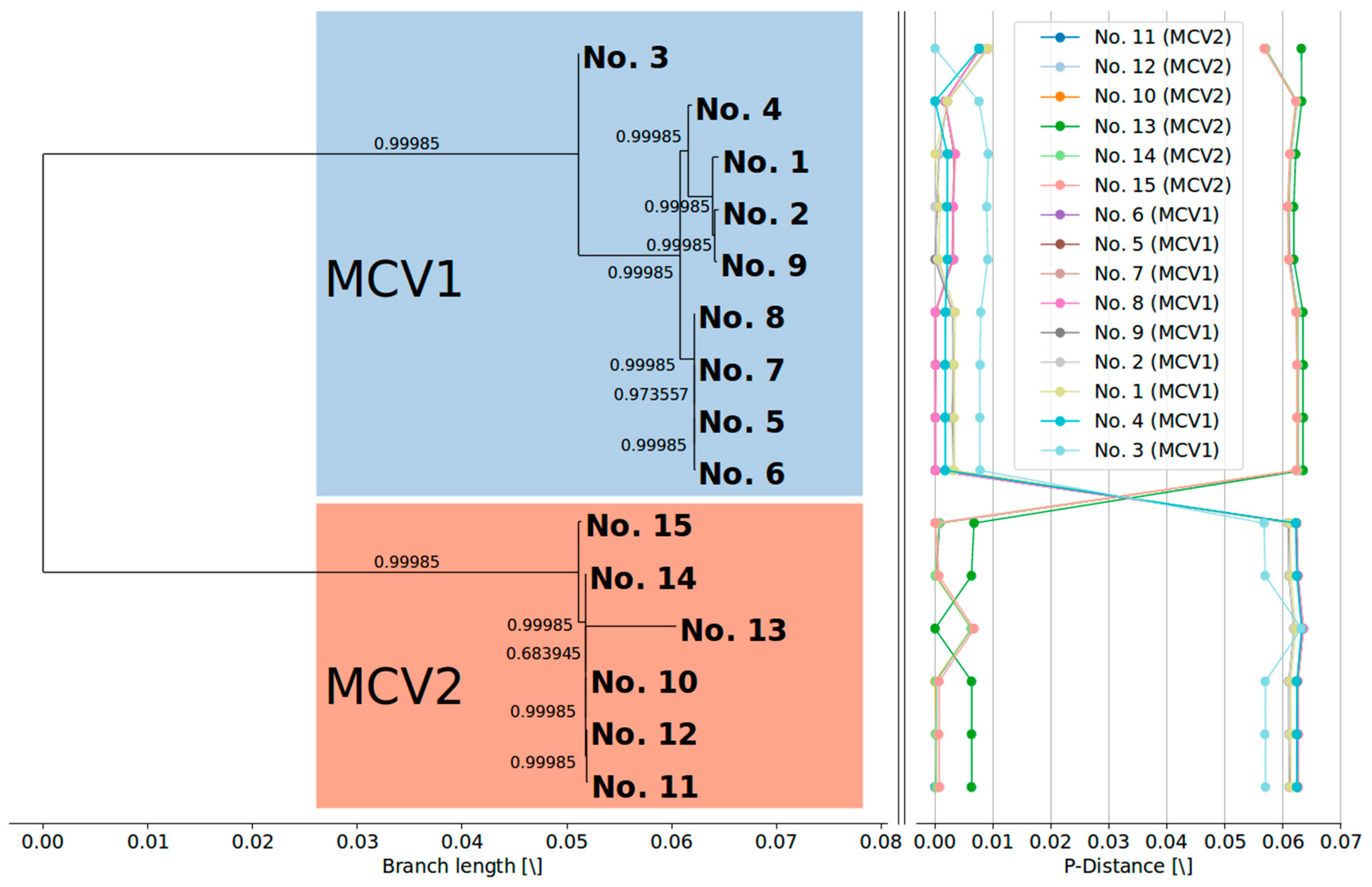
3.2. MCV1 and MCV2 Evolved from a Common Ancestor Along Divergent Evolutionary Pathways
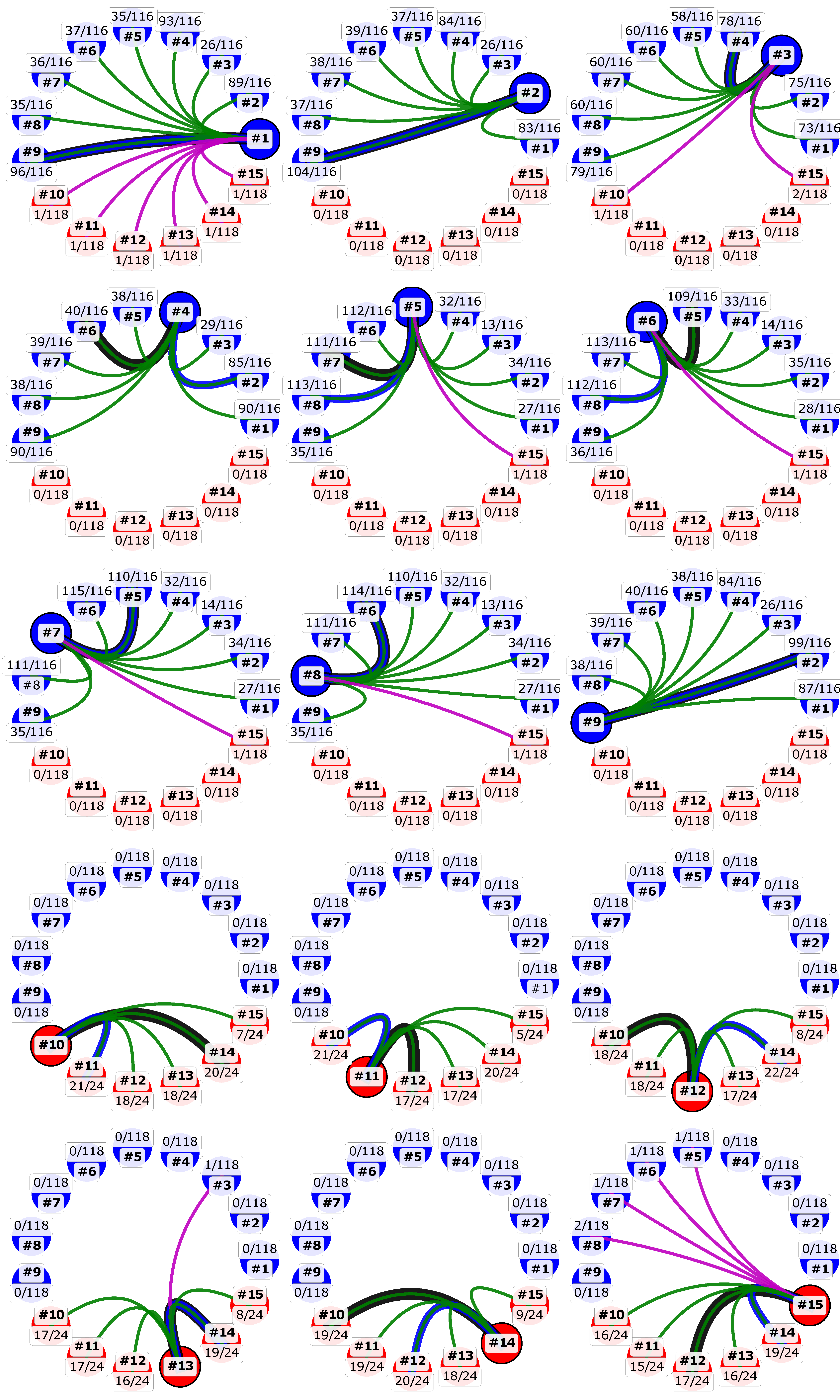
3.3. Currently Available Data Suggest that MCV1 is More Diverse than MCV2
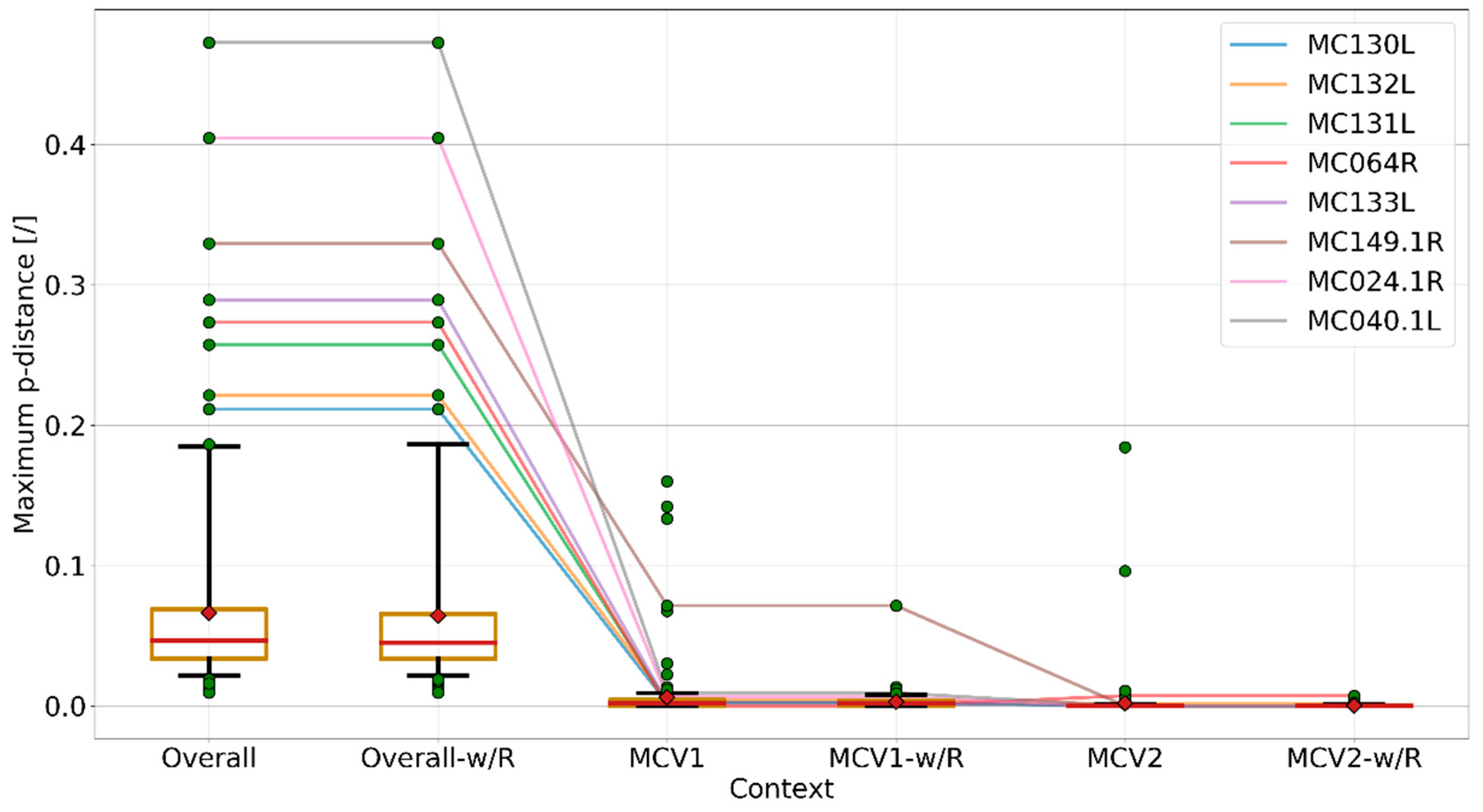
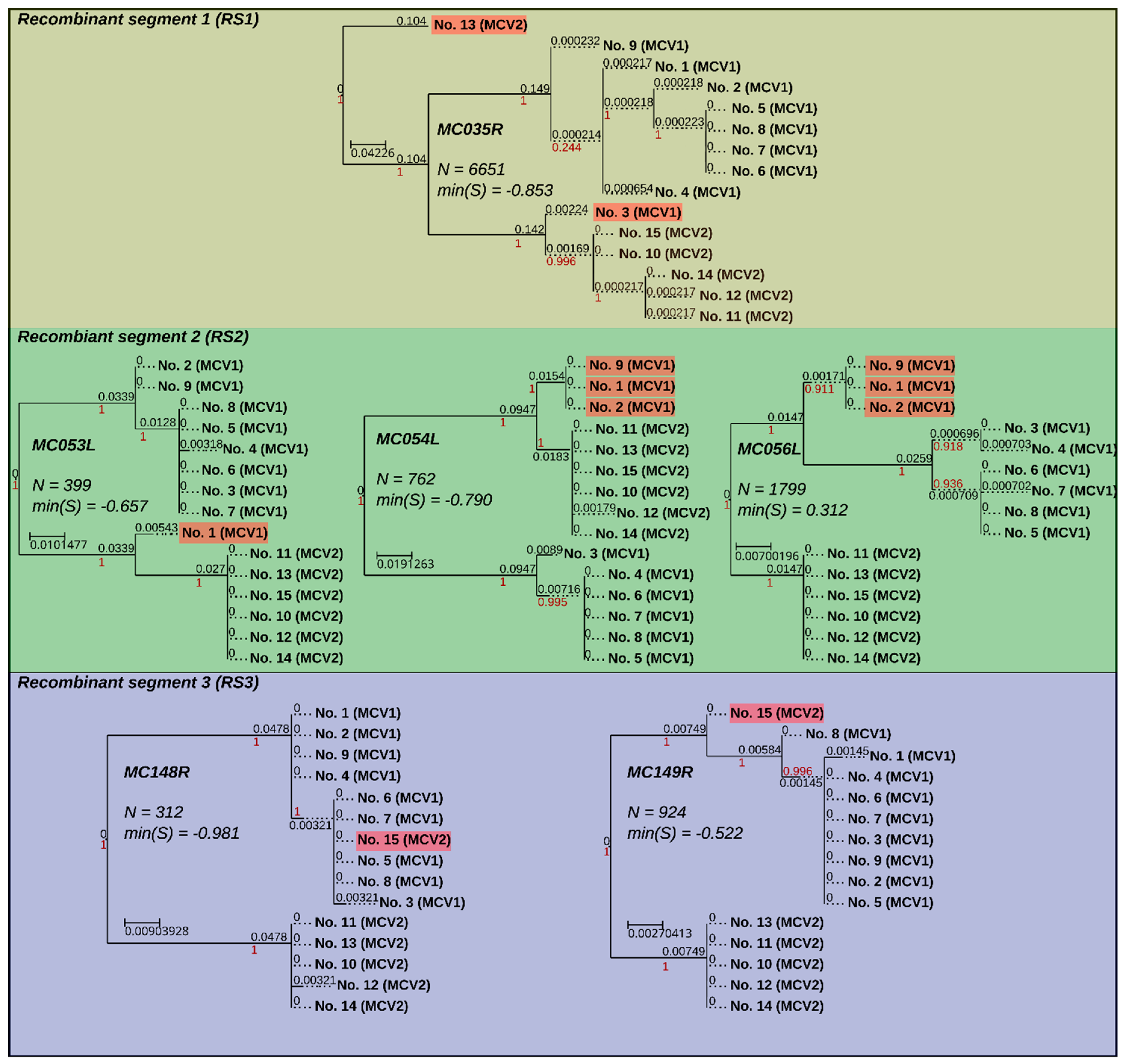
3.4. Recombination Explains Inter-Genotype Mosaicity of MCV and Anomalously High Dissimilarities Among Genes of the Same MCV Genotype
3.5. Identified Recombinant MCV Regions are Associated with Inhibition of Chemotaxis of Immune Cells and Interfering with the Host T-cell–and/or Natural Killer Cell–Related Immune Response
3.6. Higher Genomic Diversity among MCV1 Genomes in Comparison to MCV2 may be Explained by Their Preferred Hosts’ Immune Competence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, X.; Anstey, A.V.; Bugert, J.J. Molluscum contagiosum virus infection. Lancet Infect. Dis. 2013, 13, 877–888. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Bugert, J.J.; Sisler, J.R.; Koonin, E.V.; Darai, G.; Moss, B. Genome sequence of a human tumorigenic poxvirus: Prediction of specific host response-evasion genes. Science 1996, 273, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Bugert, J.J. Molluscum Contagiosum Virus. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 319–324. ISBN 9780123744104. [Google Scholar]
- Shisler, J.L. Chapter 4: Immune Evasion Strategies of Molluscum Contagiosum Virus, 1st ed.; Maramorosch-Karl, M.T., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 92, ISBN 9780128021804. [Google Scholar]
- Vermi, W.; Fisogni, S.; Salogni, L.; Schärer, L.; Kutzner, H.; Sozzani, S.; Lonardi, S.; Rossini, C.; Calzavara-Pinton, P.; Leboit, P.E.; et al. Spontaneous regression of highly immunogenic molluscum contagiosum virus (MCV)-induced skin lesions is associated with plasmacytoid dendritic cells and IFN-DC infiltration. J. Investig. Dermatol. 2011, 131, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Cotton, D.W.K.; Cooper, C.; Barrett, D.F.; Leppard, B.J. Severe atypical molluscum contagiosum in an immunocompromised host. Br. J. Dermatol. 1987, 116, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.J.; Myskowski, P.L. Molluscum contagiosum in patients with human immunodeficiency virus infection. A review of twenty-seven patients. J. Am. Acad. Dermatol. 1992, 27, 583–588. [Google Scholar] [CrossRef]
- Vora, R.V.; Pilani, A.P.; Kota, R.K. Extensive giant molluscum contagiosum in a HIV positive patient. J. Clin. Diagn. Res. 2015, 9, WD01–WD02. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.R.; Piguet, V.; Gallacher, J.; Francis, N.A. Molluscum contagiosum and associations with atopic eczema in children: A retrospective longitudinal study in primary care. Br. J. Gen. Pract. 2016, 66, e53–e58. [Google Scholar] [CrossRef] [PubMed]
- Karadag, A.S.; Karadag, R.; Bilgili, S.G.; Calka, O.; Demircan, Y.T. Giant molluscum contagiosum in an immunocompetent child. J. Pak. Med. Assoc. 2013, 63, 778–779. [Google Scholar] [CrossRef] [PubMed]
- Konya, J.; Thompson, C.H. Molluscum contagiosum virus: Antibody responses in persons with clinical lesions and seroepidemiology in a representative Australian population. J. Infect. Dis. 1999, 179, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Sherwani, S.; Farleigh, L.; Agarwal, N.; Loveless, S.; Robertson, N.; Hadaschik, E.; Schnitzler, P.; Bugert, J.J. Seroprevalence of Molluscum contagiosum virus in German and UK populations. PLoS ONE 2014, 9, e88734. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.J.; Johns, N.E.; Williams, H.C.; Bolliger, I.W.; Dellavalle, R.P.; Margolis, D.J.; Marks, R.; Naldi, L.; Weinstock, M.A.; Wulf, S.K.; et al. The global burden of skin disease in 2010: An analysis of the prevalence and impact of skin conditions. J. Investig. Dermatol. 2014, 134, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Darai, G.; Reisner, H.; Scholz, J.; Schnitzler, P.; Lorbacher de Ruiz, H. Analysis of the genome of molluscum contagiosum virus by restriction endonuclease analysis and molecular cloning. J. Med. Virol. 1986, 18, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.D.; Blake, N.W.; Archard, L.C.; Muhlemann, M.F.; Rosedale, N.; Cream, J.J. Molluscum contagiosum virus types in genital and non-genital lesions. Br. J. Dermatol. 1989, 120, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.D.; Archard, L.C. Characterisation by restriction mapping of three subtypes of molluscum contagiosum virus. J. Med. Virol. 1992, 38, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Rösen-Wolff, A.; Bugert, J.; Reisner, H.; White, M.I.; Darai, G.; Postlethwaite, R. Epidemiology of molluscum contagiosum using genetic analysis of the viral DNA. J. Med. Virol. 1989, 27, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Muraki, Y.; Yamada, M.; Hatano, Y.; Nii, S. Analysis of molluscum contagiosum virus genomes isolated in Japan. J. Med. Virol. 1995, 46, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Uemura, T.; Kawashima, M. Molecular epidemiologic analysis of Japanese patients with molluscum contagiosum. Int. J. Dermatol. 1996, 35, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, T.G.; Koonin, E.V.; Bugert, J.J.; Darai, G.; Moss, B. The genome of molluscum contagiosum virus: Analysis and comparison with other poxviruses. Virology 1997, 233, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, A.; Funes, J.; Agromayor, M.; Moratilla, M.; Varas, A.; Lopez-Estebaranz, J.; Esteban, M.; Martin-Gallardo, A. Typing of Molluscum Contagiosum Virus in Skin Lesions by Using a Simple Lysis Method. J. Med. Virol. 1996, 50, 342–349. [Google Scholar] [CrossRef]
- Thompson, C.H. Identification and Typing of Molluscum Contagiosum Virus in Clinical Specimens by Polymerase Chain Reaction. J. Med. Virol. 1997, 211, 205–211. [Google Scholar] [CrossRef]
- Trama, J.P.; Adelson, M.E.; Mordechai, E. Identification and genotyping of molluscum contagiosum virus from genital swab samples by real-time PCR and Pyrosequencing. J. Clin. Virol. 2007, 40, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Hošnjak, L.; Kocjan, B.J.; Kušar, B.; Seme, K.; Poljak, M. Rapid detection and typing of Molluscum contagiosum virus by FRET-based real-time PCR. J. Virol. Methods 2013, 187, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Rios, J.D.; Yang, Z.; Erlandson, K.J.; Cohen, J.I.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Molluscum Contagiosum Virus Transcriptome in Abortively Infected Cultured Cells and Human Skin Lesion. J. Virol. 2016, 90, 4469–4480. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Parras-Moltó, M.; López-Barrantes, O.; Belda, S.; Alejo, A. Recombination events and variability among full-length genomes of co-circulating molluscum contagiosum virus subtypes 1 and 2. J. Gen. Virol. 2017, 98, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Trčko, K.; Poljak, M.; Križmarić, M.; Miljković, J. Clinical and demographic characteristics of patients with molluscum contagiosum treated at the university dermatology clinic maribor in a 5-year period. Acta Dermatovenerol. Croat. 2016, 24, 130–136. [Google Scholar] [PubMed]
- Andrews, S. FastQC v0.10.1. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 September 2017).
- Bushnell, B. BBTools v37. Available online: https://jgi.doe.gov/data-and-tools/bbtools/ (accessed on 9 September 2017).
- Oxford Nanopore Technologies Albacore v2.0.2. Available online: https://community.nanoporetech.com/downloads (accessed on 15 June 2017).
- Wick, R. Porechop (Commit 289d5dc). Available online: https://github.com/rrwick/Porechop (accessed on 6 June 2017).
- Goodwin, S.; Gurtowski, J.; Ethe-Sayers, S.; Deshpande, P.; Schatz, M.C.; McCombie, W.R. Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015, 25, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. bioRxiv 2016, 1–35. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Vezzi, F.; Narzisi, G.; Mishra, B. Reevaluating Assembly Evaluations with Feature Response Curves: GAGE and Assemblathons. PLoS ONE 2012, 7, e52210. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.D.; Dillon, G.P.; Degrave, W.S.; Berriman, M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Mehtods to Estimate Maximum-Likelihood Phylogenies: Asessing the Performance of PhyML 2.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Lect. Math. Life Sci. 1985, 17, 57–86. [Google Scholar]
- Jones, D.; Taylor, W.; Thornton, J. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, T.E. Guide to NumPy. Methods 2010, 1, 378. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open source scientific tools for Python. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar]
- Collaboratively Developed Bioinformatics Software Scikit-Bio v0.5.2. Available online: https://www.scikit-bio.org (accessed on 9 October 2017).
- Rousseeuw, P.J. Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 1987, 20, 53–65. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P. RDP4: Instruction Manual. Ph.D. Thesis, University of Cape Town, Cape Town, South Africa, 2015. [Google Scholar]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.D.; Vasilinetc, I.; Bankevich, A.; Gurevich, A.; Krivosheeva, T.; Nurk, S.; Pham, S.; Korobeynikov, A.; Lapidus, A.; Pevzner, P.A. ExSPAnder: A universal repeat resolver for DNA fragment assembly. Bioinformatics 2014, 30, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Invergo, B.M.; Cock, P.J.A.; Chapman, B.A. Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinform. 2012, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Upton, C.; McFadden, G. Tumorigenic poxviruses: Analysis of viral DNA sequences implicated in the tumorigenicity of shope fibroma virus and malignant rabbit virus. Virology 1986, 152, 308–321. [Google Scholar] [CrossRef]
- Gershon, P.D.; Black, D.N. The nucleotide sequence around the capripoxvirus thymidine kinase gene reveals a gene shared specifically with leporipoxvirus. J. Gen. Virol. 1989, 70, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Meyer, H.; Gigante, C.M.; Gao, J.; Zhao, H.; Batra, D.; Damon, I.; Upton, C.; Li, Y. Two novel poxviruses with unusual genome rearrangements: NY_014 and Murmansk. Virus Genes 2017, 53, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Gigante, C.; Khmaladze, E.; Liu, P.; Tang, S.; Wilkins, K.; Zhao, K.; Davidson, W.; Nakazawa, Y.; Maghlakelidze, G.; et al. Genome sequences of Akhmeta virus, an early divergent old world orthopoxvirus. Viruses 2018, 10, 252. [Google Scholar] [CrossRef] [PubMed]
- Coulson, D.; Upton, C. Characterization of indels in poxvirus genomes. Virus Genes 2011, 42, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Evans, D.H. Genome Scale Patterns of Recombination between Coinfecting Vaccinia Viruses. J. Virol. 2014, 88, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Kampman, S.; Hetman, B.; Upton, C. Incongruencies in Vaccinia Virus Phylogenetic Trees. Computation 2014, 2, 182–198. [Google Scholar] [CrossRef]
- Alzhanova, D.; Hammarlund, E.; Reed, J.; Meermeier, E.; Rawlings, S.; Ray, C.A.; Edwards, D.M.; Bimber, B.; Legasse, A.; Planer, S.; et al. T Cell Inactivation by Poxviral B22 Family Proteins Increases Viral Virulence. PLoS Pathog. 2014, 10, e1004123. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, S.E.; Earl, P.L.; Minai, M.; Moore, I.; Moss, B. A homolog of the variola virus B22 membrane protein contributes to ectromelia virus pathogenicity in the mouse footpad model. Virology 2017, 501, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Moss, B. Correspondence of the functional epitopes of poxvirus and human interleukin-18-binding proteins. J. Virol. 2001, 75, 9947–9954. [Google Scholar] [CrossRef] [PubMed]
- Reading, P.C.; Smith, G.L. Vaccinia virus interleukin-18-binding protein promotes virulence by reducing gamma interferon production and natural killer and T-cell activity. J. Virol. 2003, 77, 9960–9968. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Poxvirus Protein MC132 from Molluscum Contagiosum Virus Inhibits NF-κB Activation by Targeting p65 for Degradation. J. Virol. 2015, 89, 8406–8415. [Google Scholar] [CrossRef] [PubMed]
- Brady, G.; Haas, D.A.; Farrell, P.J.; Pichlmair, A.; Bowie, A.G. Molluscum Contagiosum Virus Protein MC005 Inhibits NF-κB Activation by Targeting NEMO-Regulated IκB Kinase Activation. J. Virol. 2017, 91, e00545-17. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
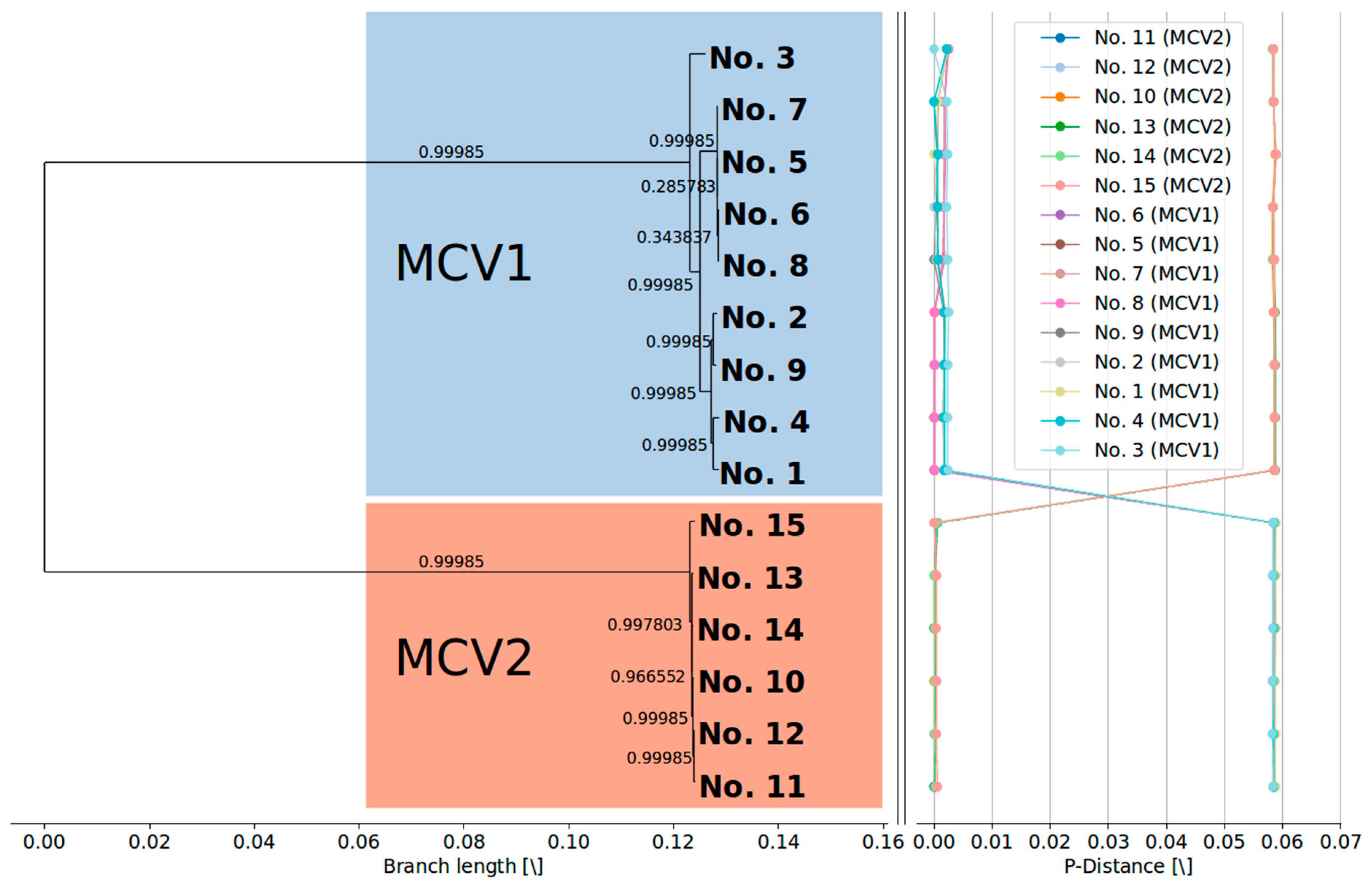{kind=link}
| No. | Viral Genotype | GenBank Acc. No. | Reference | Country of Origin | Sequencing Technique (Platform) | Assembly | Viral Load (Viral Copies/Cell) | Per-base Short Read Depth of Coverage (Mean ± SD) | Percentage of Mapped Short Reads (%) | Genome Length (nt) | ITR Length (nt) | Number of Annotated Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MCV1 | U60315 | Senkevich et al. [2] | Unknown | Applied Biosystems AB373A (primer-walking) | / | / | / | / | 190,289 | 4711 | 178 |
| 2 | MCV1 | KY040275 | López-Bueno et al. [26] | Spain | Illumina MiSeq (2 × 300 nt) | Short-read | / | / | / | 188,253 | 3821 | 181 |
| 3 | MCV1 | KY040276 | López-Bueno et al. [26] | Spain | Illumina MiSeq (2 × 300 nt) | Short-read | / | / | / | 189,098 | 4252 | 179 |
| 4 | MCV1 | KY040277 | López-Bueno et al. [26] | Spain | Illumina MiSeq (2 × 300 nt) | Short-read | / | / | / | 188,458 | 3758 | 179 |
| 5 | MCV1 | MH320553 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt), ONT | Hybrid | 4237 | 1772.92 ± 282.67 | 12.30 | 187,558 | 3519 | 177 |
| 6 | MCV1 | MH320552 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt), ONT | Hybrid | 2527 | 3864.52 ± 526.58 | 26.11 | 187,884 | 3651 | 176 |
| 7 | MCV1 | MH320547 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt) | Short-read | 1021 | 2243.29 ± 750.52 | 18.37 | 187,826 | 3559 | 177 |
| 8 | MCV1 | MH320555 | This study | Slovenia | Illumina HiSeq2000 (2 × 150 nt, 2 × 250 nt), ONT | Hybrid | 546,855 | 635.62 ± 208.74 | 87.98 | 189,292 | 4354 | 176 |
| 9 | MCV1 | MH320554 | This study | Slovenia | Illumina HiSeq2000 (2 × 150 nt; 2 × 250 nt), ONT | Hybrid | 40,351 | 581.67 ± 134.87 | 44.27 | 196,781 | 7975 | 175 |
| 10 | MCV2 | KY040274 | López-Bueno et al. [26] | Spain | Illumina MiSeq (2 × 300 nt) | Short-read | / | / | / | 192,183 | 4086 | 170 |
| 11 | MCV2 | MH320550 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt), ONT | Hybrid | 26,717 | 2913.56 ± 417.96 | 18.53 | 196,206 | 7762 | 170 |
| 12 | MCV2 | MH320548 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt) | Short-read | 5226 | 5270.58 ± 1499.21 | 27.27 | 190,319 | 4937 | 170 |
| 13 | MCV2 | MH320556 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt) | Short-read | 4573 | 5861.15 ± 622.65 | 39.18 | 189,257 | 4319 | 170 |
| 14 | MCV2 | MH320551 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt), ONT | Hybrid | 1828 | 3543.65 ± 546.386 | 24.24 | 192,156 | 5979 | 170 |
| 15 | MCV2 | MH320549 | This study | Slovenia | Illumina HiSeq4000 (2 × 150 nt), ONT | Hybrid | 8727 | 1912.23 ± 416.643 | 13.30 | 193,271 | 6432 | 170 |
| Gene | Missing in Genomes (Count) | Missing in Genomes (Sequence No.) | Function/Homologues/Reference |
|---|---|---|---|
| MC001R | 3 | 7, 8, 9 | Predicted non-globular protein/MC164L/Senkevich et al. [20] |
| MC006.1R | 6 | 10 *, 11, 12, 13, 14, 15 | Unknown/ /Senkevich et al. [20] |
| MC009.1R | 2 | 1 *, 4 * | Predicted non-globular protein/ /Senkevich et al. [20] |
| MC009.2R | 1 | 1 * | Predicted non-globular protein/ /Senkevich et al. [20] |
| MC017.1L | 12 | 3 *, 5, 6, 7, 8, 9, 8, 10 *, 11, 12, 13, 15 | Predicted non-globular protein/ /Senkevich et al. [20] |
| MC022.1L | 6 | 3 *, 5, 6, 7, 8, 9 | Unknown/ /Senkevich et al. [20] |
| MC042.1R | 8 | 1 *, 2 *, 10 *, 11, 12, 13, 14, 15 | Predicted structural protein/ /Senkevich et al. [20] |
| MC052R | 6 | 10 *, 11, 12, 13, 14, 15 | Unknown/ /Senkevich et al. [20] |
| MC053.1R | 13 | 3 *, 4 *, 5, 6, 7, 8, 9, 10 *, 11, 12, 13, 14, 15 | Predicted structural protein/ /Senkevich et al. [20] |
| MC053.2R | 7 | 4 *, 10 *, 11, 12, 13, 14, 15 | Predicted C-terminal transmembrane helix/ /Senkevich et al. [20] |
| MC055R | 6 | 10 *, 11, 12, 13, 14, 15 | Unknown/ /Senkevich et al. [20] |
| MC144R | 6 | 10 *, 11, 12, 13, 14, 15 | Predicted long non-globular protein/ /Senkevich et al. [20] |
| MC145.1R | 1 | 1 * | Predicted non-globular protein/ /Senkevich et al. [20] |
| MC147R | 6 | 10 *, 11, 12, 13, 14, 15 | Unknown/ /Senkevich et al. [20] |
| MC150R | 7 | 6, 10 *, 11, 12, 13, 14, 15 | Unknown/ /Senkevich et al. [20] |
| MC152.1R | 1 | 3 * | Unknown/ /Senkevich et al. [20] |
| MC156R | 7 | 6, 10 *, 11, 12, 13, 14, 15 | Predicted peptide, putative secreted protein/ /NCBI Gene database |
| MC164L | 9 | 5, 8, 9, 10 *, 11, 12, 13, 14, 15 | Predicted non-globular protein/MC001R/Senkevich et al. [20] |
| Mean p-Distances | GC Content | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample vs. All | Intra-Genotype | ||||||||
| Number | Viral Genotype | Genome | Genome (Balancing) | Consensus Genes | Consensus Genes (Balancing) | Genome | Consensus Genes | Genotype | Consensus Genes |
| 1 | MCV1 | 0.02821 ± 0.02885 | 0.03500 ± 3.3 × 10−4 | 0.02490 ± 0.02582 | 0.03100 ± 3.0 × 10−4 | 0.002909 ± 2.546 × 10−3 | 0.002314 ± 2.521 × 10−3 | 0.6336 | 0.6435 |
| 2 | MCV1 | 0.02793 ± 0.02877 | 0.03471 ± 3.2 × 10−4 | 0.02484 ± 0.02595 | 0.03100 ± 3.0 × 10−4 | 0.002730 ± 2.493 × 10−3 | 0.002158 ± 2.497 × 10−3 | 0.6342 | 0.6333 |
| 3 | MCV1 | 0.02954 ± 0.02465 | 0.03535 ± 9 × 10−5 | 0.02690 ± 0.02138 | 0.03193 ± 5 × 10−5 | 0.007318 ± 2.658 × 10−3 | 0.007500 ± 2.675 × 10−3 | 0.6338 | 0.6430 |
| 4 | MCV1 | 0.02827 ± 0.02966 | 0.03526 ± 2.5 × 10−4 | 0.02504 ± 0.02642 | 0.03127 ± 3.0 × 10−4 | 0.002317 ± 1.959 × 10−3 | 0.001969 ± 2.675 × 10−3 | 0.6345 | 0.6433 |
| 5 | MCV1 | 0.02822 ± 0.02991 | 0.03527 ± 3.2 × 10−4 | 0.02497 ± 0.02657 | 0.03123 ± 3.0 × 10−4 | 0.002107 ± 2.390 × 10−3 | 0.001795 ± 2.418 × 10−3 | 0.6341 | 0.6431 |
| 6 | MCV1 | 0.02824 ± 0.02987 | 0.03528 ± 3.2 × 10−4 | 0.02500 ± 0.02660 | 0.03127 ± 3.0 × 10−4 | 0.002152 ± 2.411 × 10−3 | 0.001794 ± 2.417 × 10−3 | 0.6339 | 0.6432 |
| 7 | MCV1 | 0.02824 ± 0.02989 | 0.03529 ± 3.2 × 10−4 | 0.02512 ± 0.02658 | 0.03138 ± 3.0 × 10−4 | 0.002140 ± 2.408 × 10−3 | 0.001919 ± 2.440 × 10−3 | 0.6340 | 0.6431 |
| 8 | MCV1 | 0.02823 ± 0.02982 | 0.03526 ± 3.3 × 10−4 | 0.02496 ± 0.02657 | 0.03122 ± 3.0 × 10−4 | 0.002181 ± 2.445 × 10−3 | 0.001785 ± 2.412 × 10−3 | 0.6332 | 0.6430 |
| 9 | MCV1 | 0.02798 ± 0.02881 | 0.03477 ± 3.3 × 10−4 | 0.02482 ± 0.02598 | 0.03094 ± 3.0 × 10−4 | 0.002736 ± 2.547 × 10−3 | 0.002122 ± 2.508 × 10−3 | 0.6312 | 0.6434 |
| 10 | MCV2 | 0.03999 ± 0.02877 | 0.03415 ± 2.3 × 10−4 | 0.03552 ± 0.02551 | 0.03035 ± 2.0 × 10−4 | 0.001233 ± 2.271 × 10−3 | 0.001168 ± 2.410 × 10−3 | 0.6432 | 0.6524 |
| 11 | MCV2 | 0.04005 ± 0.02877 | 0.03421 ± 2.3 × 10−4 | 0.03557 ± 0.02554 | 0.03039 ± 2.0 × 10−4 | 0.001263 ± 2.256 × 10−3 | 0.001173 ± 2.408 × 10−3 | 0.6403 | 0.6524 |
| 12 | MCV2 | 0.04002 ± 0.02881 | 0.03418 ± 2.4 × 10−4 | 0.03557 ± 0.02554 | 0.03040 ± 2.0 × 10−4 | 0.001223 ± 2.274 × 10−3 | 0.001177 ± 2.415 × 10−3 | 0.6438 | 0.6523 |
| 13 | MCV2 | 0.04271 ± 0.02710 | 0.03721 ± 9 × 10−5 | 0.03866 ± 0.02389 | 0.03380 ± 7 × 10−5 | 0.005307 ± 2.379 × 10−3 | 0.005506 ± 2.464 × 10−3 | 0.6441 | 0.6518 |
| 14 | MCV2 | 0.04002 ± 0.02880 | 0.03418 ± 2.4 × 10−4 | 0.03557 ± 0.02554 | 0.03039 ± 2.0 × 10−4 | 0.001231 ± 2.263 × 10−3 | 0.001165 ± 2.409 × 10−3 | 0.6424 | 0.6523 |
| 15 | MCV2 | 0.04004 ± 0.02850 | 0.03426 ± 2.3 × 10−4 | 0.03560 ± 0.02539 | 0.03045 ± 2.0 × 10−4 | 0.001580 ± 2.314 × 10−3 | 0.001365 ± 2.4426 × 10−3 | 0.6414 | 0.6523 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorec, T.M.; Kutnjak, D.; Hošnjak, L.; Kušar, B.; Trčko, K.; Kocjan, B.J.; Li, Y.; Križmarić, M.; Miljković, J.; Ravnikar, M.; et al. New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences. Viruses 2018, 10, 586. https://doi.org/10.3390/v10110586
Zorec TM, Kutnjak D, Hošnjak L, Kušar B, Trčko K, Kocjan BJ, Li Y, Križmarić M, Miljković J, Ravnikar M, et al. New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences. Viruses. 2018; 10(11):586. https://doi.org/10.3390/v10110586
Chicago/Turabian StyleZorec, Tomaž M., Denis Kutnjak, Lea Hošnjak, Blanka Kušar, Katarina Trčko, Boštjan J. Kocjan, Yu Li, Miljenko Križmarić, Jovan Miljković, Maja Ravnikar, and et al. 2018. "New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences" Viruses 10, no. 11: 586. https://doi.org/10.3390/v10110586
APA StyleZorec, T. M., Kutnjak, D., Hošnjak, L., Kušar, B., Trčko, K., Kocjan, B. J., Li, Y., Križmarić, M., Miljković, J., Ravnikar, M., & Poljak, M. (2018). New Insights into the Evolutionary and Genomic Landscape of Molluscum Contagiosum Virus (MCV) based on Nine MCV1 and Six MCV2 Complete Genome Sequences. Viruses, 10(11), 586. https://doi.org/10.3390/v10110586