Phylogenetic Analysis and Characterization of a Sporadic Isolate of Equine Influenza A H3N8 from an Unvaccinated Horse in 2015


Abstract
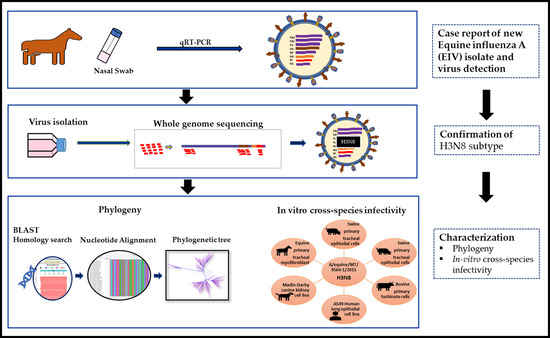
1. Introduction
2. Materials and Methods
2.1. Case Description
2.2. Whole Genome Sequencing and Phylogenetic Analysis
2.3. Virus and Cell Culture
2.4. Replication Kinetics
3. Results
3.1. BLAST Analysis
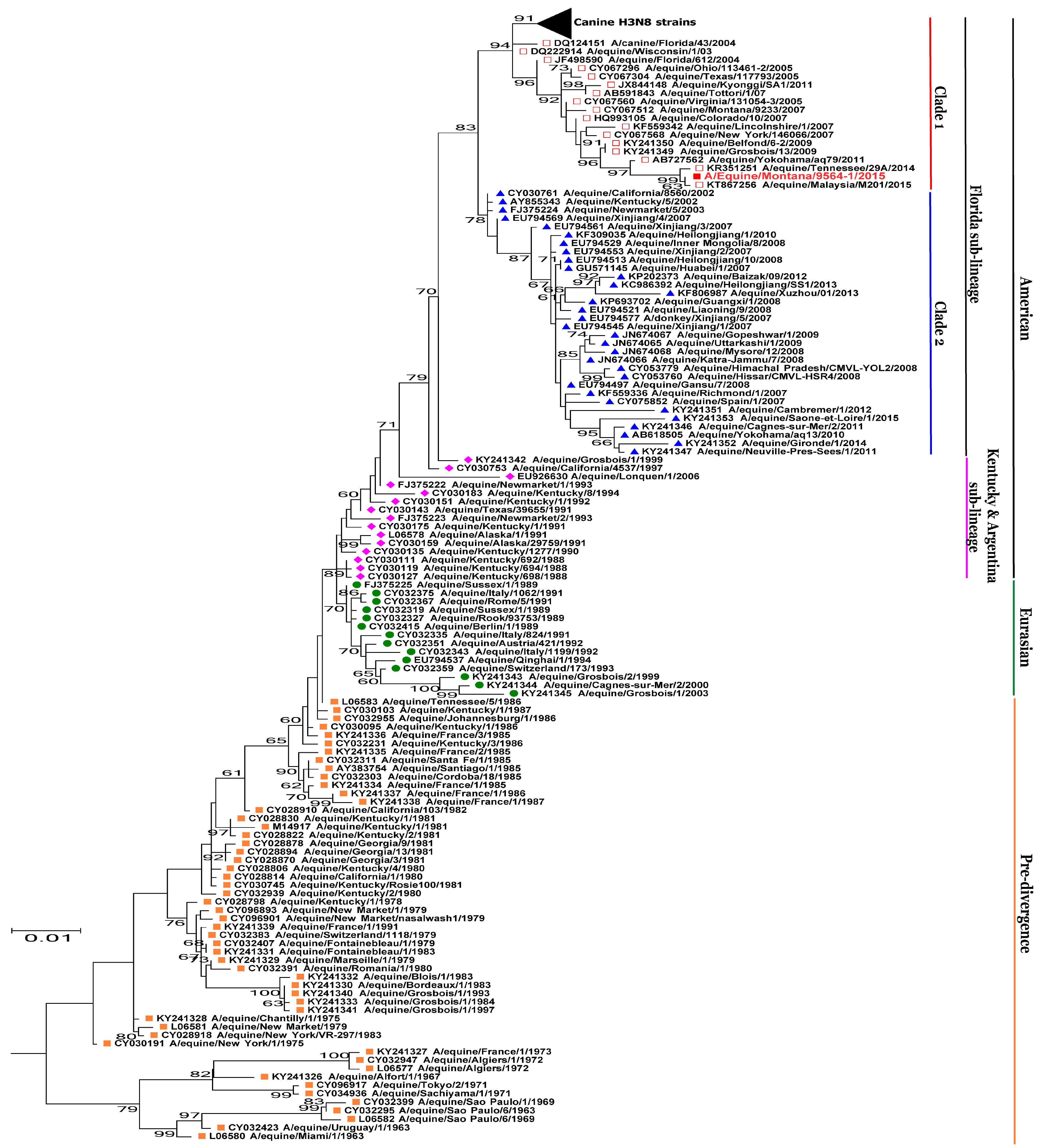
3.2. Phylogenetic Analysis
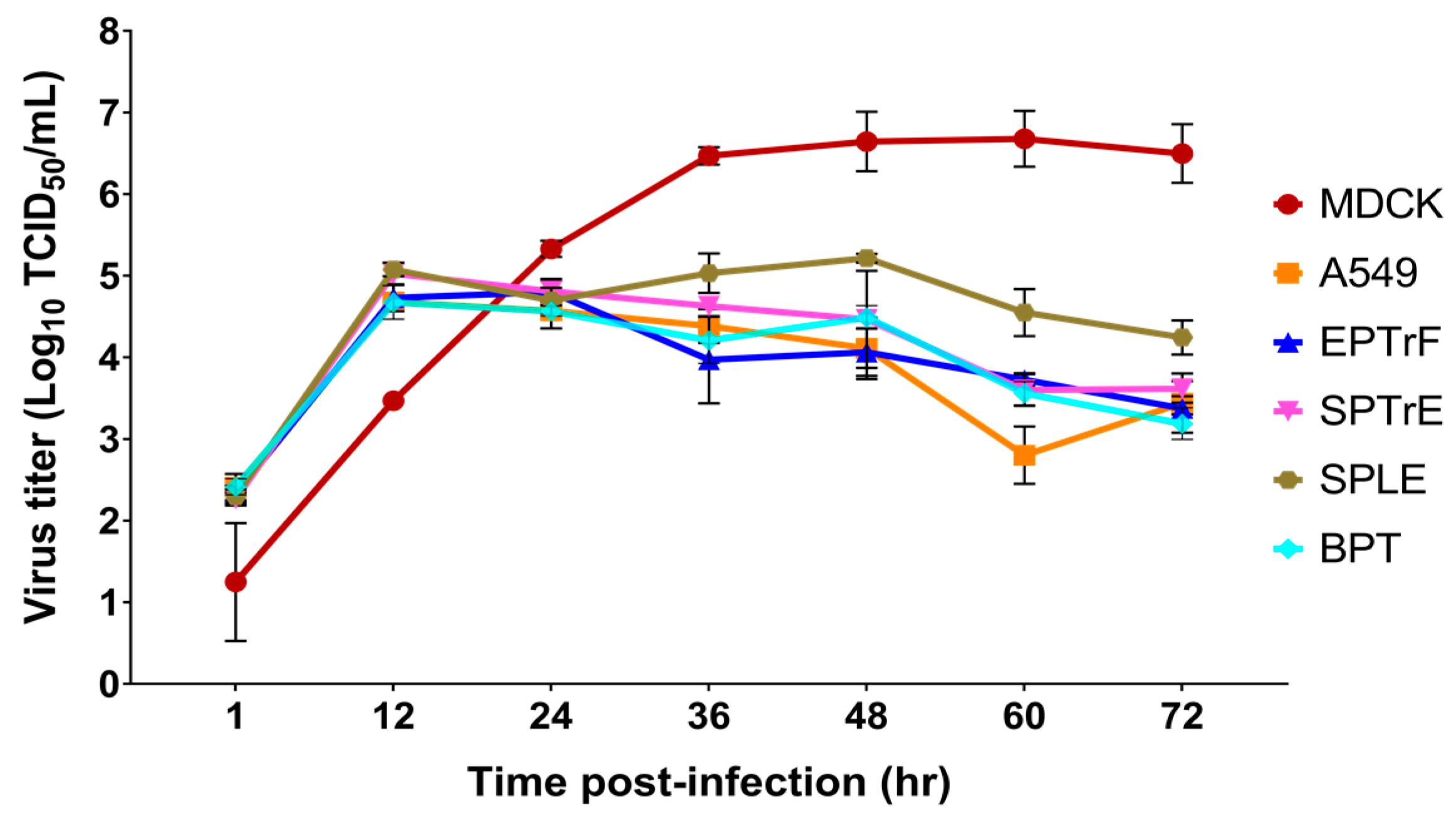
3.3. Viral Replication Kinetics
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lai, A.C.K.; Chambers, T.M.; Holland, J.R.E.; Morley, P.S.; Haines, D.M.; Townsend, H.G.G.; Barrandeguy, M. Diverged evolution of recent equine-2 influenza (H3N8) viruses in the Western Hemisphere. Arch. Virol. 2001, 146, 1063–1074. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, J.; Newton, R.; Wood, J.; Geraghty, B.; Ellis, R. Equine influenza in donkeys in the new forest. Vet. Rec. 2000, 147, 400. [Google Scholar] [PubMed]
- Mumford, E.L.; Traub-Dargatz, J.L.; Salman, M.D.; Collins, J.K.; Getzy, D.M.; Carman, J. Monitoring and detection of acute viral respiratory tract disease in horses. J. Am. Vet. Med. Assoc. 1998, 213, 385–390. [Google Scholar] [PubMed]
- Timoney, P.J. Equine influenza. Comp. Immunol. Microbiol. Infect. Dis. 1996, 19, 205–211. [Google Scholar] [CrossRef]
- Wilson, W.D. Equine influenza. Vet. Clin. N. Am. Equine Pract. 1993, 9, 257–282. [Google Scholar] [CrossRef]
- Cullinane, A.; Newton, J.R. Equine influenza—A global perspective. Vet. Microbiol. 2013, 167, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Alves Beuttemmuller, E.; Woodward, A.; Rash, A.; Dos Santos Ferraz, L.E.; Fernandes Alfieri, A.; Alfieri, A.A.; Elton, D. Characterisation of the epidemic strain of H3N8 equine influenza virus responsible for outbreaks in South America in 2012. Virol. J. 2016, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, R.H.; Kehler, W.H.; Henthorne, J.C. The 1963 equine influenza epizootic. J. Am. Vet. Med. Assoc. 1963, 143, 1108–1110. [Google Scholar] [PubMed]
- Scholtens, R.G.; Steele, J.H.; Dowdle, W.R.; Yarbrough, W.B.; Robinson, R.Q. U.S. Epizootic of equine influenza, 1963. Public Health Rep. 1964, 79, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sovinova, O.; Tumova, B.; Pouska, F.; Nemec, J. Isolation of a virus causing respiratory disease in horses. Acta Virol. 1958, 2, 52–61. [Google Scholar] [PubMed]
- Webster, R.G. Are equine 1 influenza viruses still present in horses? Equine Vet. J. 1993, 25, 537–538. [Google Scholar] [CrossRef] [PubMed]
- The ecology of influenza viruses: A who memorandum. Bull. World Health Organ. 1981, 59, 869–873.
- Waddell, G.H.; Teigland, M.B.; Sigel, M.M. A new influenza virus associated with equine respiratory disease. J. Am. Vet. Med. Assoc. 1963, 143, 587–590. [Google Scholar] [PubMed]
- Wilson, J.C.; Bryans, J.T.; Doll, E.R. Recovery of influenza virus from horses in the equine influenza epizootic of 1963. Am. J. Vet. Res. 1965, 26, 1466–1468. [Google Scholar] [PubMed]
- Daly, J.M.; Lai, A.C.; Binns, M.M.; Chambers, T.M.; Barrandeguy, M.; Mumford, J.A. Antigenic and genetic evolution of equine H3N8 influenza a viruses. J. Gen. Virol. 1996, 77, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Murcia, P.R.; Wood, J.L.; Holmes, E.C. Genome-scale evolution and phylodynamics of equine H3N8 influenza a virus. J. Virol. 2011, 85, 5312–5322. [Google Scholar] [CrossRef] [PubMed]
- Rash, A.; Morton, R.; Woodward, A.; Maes, O.; McCauley, J.; Bryant, N.; Elton, D. Evolution and divergence of H3N8 equine influenza viruses circulating in the United Kingdom from 2013 to 2015. Pathogens 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fougerolle, S.; Legrand, L.; Lecouturier, F.; Sailleau, C.; Paillot, R.; Hans, A.; Pronost, S. Genetic evolution of equine influenza virus strains (H3N8) isolated in france from 1967 to 2015 and the implications of several potential pathogenic factors. Virology 2017, 505, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Bountouri, M.; Fragkiadaki, E.; Ntafis, V.; Kanellos, T.; Xylouri, E. Phylogenetic and molecular characterization of equine H3N8 influenza viruses from greece (2003 and 2007): Evidence for reassortment between evolutionary lineages. Virol. J. 2011, 8, 350. [Google Scholar] [CrossRef] [PubMed]
- Perglione, C.O.; Gildea, S.; Rimondi, A.; Mino, S.; Vissani, A.; Carossino, M.; Cullinane, A.; Barrandeguy, M. Epidemiological and virological findings during multiple outbreaks of equine influenza in South America in 2012. Influenza Other Respir. Viruses 2016, 10, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Olguin Perglione, C.; Golemba, M.D.; Torres, C.; Barrandeguy, M. Molecular epidemiology and spatio-temporal dynamics of the H3N8 equine influenza virus in South America. Pathogens 2016, 5. [Google Scholar] [CrossRef]
- Crispe, E.; Finlaison, D.S.; Hurt, A.C.; Kirkland, P.D. Infection of dogs with equine influenza virus: Evidence for transmission from horses during the Australian outbreak. Aust. Vet. J. 2011, 89 (Suppl. 1), 27–28. [Google Scholar] [CrossRef] [PubMed]
- Bryant, N.A.; Rash, A.S.; Russell, C.A.; Ross, J.; Cooke, A.; Bowman, S.; MacRae, S.; Lewis, N.S.; Paillot, R.; Zanoni, R.; et al. Antigenic and genetic variations in european and North American equine influenza virus strains (H3N8) isolated from 2006 to 2007. Vet. Microbiol. 2009, 138, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, T.; Nemoto, M.; Bannai, H.; Tsujimura, K.; Kondo, T.; Matsumura, T.; Gildea, S.; Cullinane, A. Assessment of antigenic difference of equine influenza virus strains by challenge study in horses. Influenza Other Respir. Viruses 2016, 10, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Anderson, B.D.; Daramragchaa, U.; Chuluunbaatar, M.; Gray, G.C. A review of evidence that equine influenza viruses are zoonotic. Pathogens 2016, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Paillot, R. A systematic review of recent advances in equine influenza vaccination. Vaccines (Basel) 2014, 2, 797–831. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Allen, R.C.; Baguelin, M.; Hampson, K.; Baillie, G.J.; Elton, D.; Newton, J.R.; Kellam, P.; Wood, J.L.; Holmes, E.C.; et al. Transmission of equine influenza virus during an outbreak is characterized by frequent mixed infections and loose transmission bottlenecks. PLoS Pathog. 2012, 8, e1003081. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, P.D.; Finlaison, D.S.; Crispe, E.; Hurt, A.C. Influenza virus transmission from horses to dogs, australia. Emerg. Infect. Dis. 2010, 16, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Marshall, J.F.; Morrell, J.; Robb, D.; McCauley, J.W.; Perez, D.R.; Parrish, C.R.; Murcia, P.R. Infection and pathogenesis of canine, equine, and human influenza viruses in canine tracheas. J. Virol. 2014, 88, 9208–9219. [Google Scholar] [CrossRef] [PubMed]
- Crawford, P.C.; Dubovi, E.J.; Castleman, W.L.; Stephenson, I.; Gibbs, E.P.; Chen, L.; Smith, C.; Hill, R.C.; Ferro, P.; Pompey, J.; et al. Transmission of equine influenza virus to dogs. Science 2005, 310, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.M.; Blunden, A.S.; Macrae, S.; Miller, J.; Bowman, S.J.; Kolodziejek, J.; Nowotny, N.; Smith, K.C. Transmission of equine influenza virus to English foxhounds. Emerg. Infect. Dis. 2008, 14, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Murcia, P.R.; Wood, J.L. Equine influenza virus: A jumping virus that races with thoroughbred horses and greyhounds. Vet. J. 2011, 189, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Holland, R.E., Jr.; McCoy, M.H.; Donofrio-Newman, J.; Vickers, M.L.; Chambers, T.M. Infectivity of equine H3N8 influenza virus in bovine cells and calves. Influenza Other Respir. Viruses 2010, 4, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wang, L.; Fu, X.; He, S.; Hong, M.; Zhou, P.; Lai, A.; Gray, G.; Li, S. Equine influenza a(H3N8) virus infection in cats. Emerg. Infect. Dis. 2014, 20, 2096–2099. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Zhou, H.; Jiang, T.; Li, C.; Zhang, A.; Guo, X.; Zou, W.; Chen, H.; Jin, M. Isolation and molecular characterization of equine H3N8 influenza viruses from pigs in China. Arch. Virol. 2009, 154, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Kasel, J.A.; Alford, R.H.; Knight, V.; Waddell, G.H.; Sigel, M.M. Experimental infection of human volunteers with equine influenza virus. Nature 1965, 206, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Minuse, E.; McQueen, J.L.; Davenport, F.M.; Francis, T., Jr. Studies of antibodies to 1956 and 1963 equine influenza viruses in horses and man. J. Immunol. 1965, 94, 563–566. [Google Scholar] [PubMed]
- Morens, D.M.; Taubenberger, J.K. Historical thoughts on influenza viral ecosystems, or behold a pale horse, dead dogs, failing fowl, and sick swine. Influenza Other Respir. Viruses 2010, 4, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.R.; Heil, G.L.; Chambers, T.M.; Capuano, A.; White, S.K.; Gray, G.C. Serological evidence of equine influenza infections among persons with horse exposure, Iowa. J. Clin. Virol. 2015, 67, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Van Oirschot, J.T.; Bruin, G.; de Boer-Luytze, E.; Smolders, G. Maternal antibodies against equine influenza virus in foals and their interference with vaccination. Zentralbl. Veterinarmed. B 1991, 38, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Van Oirschot, J.T.; Masurel, N.; Huffels, A.D.; Anker, W.J. Equine influenza in The Netherlands during the winter of 1978–1979; antigenic drift of the A-equi 2 virus. Vet. Q. 1981, 3, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Paillot, R.; Rash, N.L.; Garrett, D.; Prowse-Davis, L.; Montesso, F.; Cullinane, A.; Lemaitre, L.; Thibault, J.C.; Wittreck, S.; Dancer, A. How to meet the last OIE expert surveillance panel recommendations on equine influenza (EI) vaccine composition: A review of the process required for the recombinant canarypox-based EI vaccine. Pathogens 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Office International des Epizooties (OIE). Conclusions and Recommendations-OIE Expert Surveillance Panel on Equine Influenza Vaccine Composition, OIE Headquarters, 22 March 2017 (accessed on 10 October 2017); Office International des Epizooties: Paris, France, 2017. [Google Scholar]
- Elton, D.; Cullinane, A. Equine influenza: Antigenic drift and implications for vaccines. Equine Vet. J. 2013, 45, 768–769. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Bolotov, P.; Dernovoy, D.; Kiryutin, B.; Zaslavsky, L.; Tatusova, T.; Ostell, J.; Lipman, D. The influenza virus resource at the national center for biotechnology information. J. Virol. 2008, 82, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the Orthomyxoviridae family. MBio 2014, 5, e00031-14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed blastn: An accelerated megablast search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Oliver, J.L.; Marin, A.; Medina, J.R. The general stochastic model of nucleotide substitution. J. Theor. Biol. 1990, 142, 485–501. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Trop. Med. Hyg. 1938, 27, 493–497. [Google Scholar]
- Chang, S.; Zhang, J.; Liao, X.; Zhu, X.; Wang, D.; Zhu, J.; Feng, T.; Zhu, B.; Gao, G.F.; Wang, J.; et al. Influenza virus database (IVDB): An integrated information resource and analysis platform for influenza virus research. Nucleic Acids Res. 2007, 35, D376–D380. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.C.; Rogers, K.M.; Glaser, A.; Tudor, L.; Chambers, T. Alternate circulation of recent equine-2 influenza viruses (H3N8) from two distinct lineages in the United States. Virus Res. 2004, 100, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, A.J. Equine Influenza in South Africa, 2003 outbreak. In Proceedings of the 9th International Congress of World Equine Veterinary Association, Marrakech, Morocco, 22–26 January 2016. [Google Scholar]
- Paillot, R.; El-Hage, C.M. The use of a recombinant canarypox-based equine influenza vaccine during the 2007 Australian outbreak: A systematic review and summary. Pathogens 2016, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Pusterla, N.; Kass, P.H.; Mapes, S.; Wademan, C.; Akana, N.; Barnett, C.; MacKenzie, C.; Vaala, W. Voluntary surveillance program for equine influenza virus in the United States from 2010 to 2013. J. Vet. Intern. Med. 2015, 29, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Nath, D.M.; Minocha, H.C. Replication of swine and equine influenza viruses in canine kidney cells. Am. J. Vet. Res. 1977, 38, 1059–1061. [Google Scholar] [PubMed]
- Patrono, L.V.; Bonfante, F.; Zanardello, C.; Terregino, C.; Capua, I.; Murcia, P.R. Phylogenetically distinct equine influenza viruses show different tropism for the swine respiratory tract. J. Gen. Virol. 2015, 96, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.H.; Gonzalez, G.; Deng, L.; Yu, H.; Tse, V.L.; Huang, L.; Huang, K.; Wasik, B.R.; Zhou, B.; Wentworth, D.E.; et al. Equine and canine influenza H3N8 viruses show minimal biological differences despite phylogenetic divergence. J. Virol. 2015, 89, 6860–6873. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Kahn, R.E.; Richt, J.A. The pig as a mixing vessel for influenza viruses: Human and veterinary implications. J. Mol. Genet. Med. 2009, 3, 158–166. [Google Scholar] [CrossRef]
- Seitz, C.; Frensing, T.; Höper, D.; Kochs, G.; Reichl, U. High yields of influenza a virus in madin–darby canine kidney cells are promoted by an insufficient interferon-induced antiviral state. J. Gen. Virol. 2010, 91, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Lu, J.; Yu, R.; Wang, X.; Wang, T.; Dong, F.; Peng, B.; Wu, W.; Liu, H.; Geng, Y.; et al. Preliminary proteomic analysis of A549 cells infected with avian influenza virus H7N9 and influenza a virus H1N1. PLoS ONE 2016, 11, e0156017. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.O.; Stertz, S. Measuring attachment and internalization of influenza a virus in a549 cells by flow cytometry. J. Vis. Exp. 2015, e53372. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Yashiro, M.; Ogawa, H.; Namba, H.; Nosaka, N.; Fujii, Y.; Morishima, T.; Tsukahara, H.; Yamada, M. Metabolic pathway catalyzed by vanin-1 pantetheinase plays a suppressive role in influenza virus replication in human alveolar epithelial A549 cells. Biochem. Biophys. Res. Commun. 2017, 489, 466–471. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession No. | Viruses with Highest % of Nucleotide Identity | Percent Identity |
|---|---|---|---|
| PB2 | MF599501.1 | A/equine/Malaysia/M201/2015 (H3N8) | 99.83 |
| PB1 | KR351247.1 | A/equine/Tennessee/29A/2014 (H3N8) | 99.78 |
| PA | KR351248.1 | A/equine/Tennessee/29A/2014 (H3N8) | 99.81 |
| HA | KR351249.1 | A/equine/Tennessee/29A/2014 (H3N8) | 99.54 |
| NP | KR351250.1 | A/equine/Tennessee/29A/2014 (H3N8) | 99.87 |
| NA | KT867256.1 | A/equine/Malaysia/M201/2015 (H3N8) | 99.93 |
| M | KR351252.1 | A/equine/Tennessee/29A/2014 (H3N8) | 99.8 |
| NS | MF599503.1 | A/equine/Malaysia/M201/2015 (H3N8) | 100.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sreenivasan, C.C.; Jandhyala, S.S.; Luo, S.; Hause, B.M.; Thomas, M.; Knudsen, D.E.B.; Leslie-Steen, P.; Clement, T.; Reedy, S.E.; Chambers, T.M.; et al. Phylogenetic Analysis and Characterization of a Sporadic Isolate of Equine Influenza A H3N8 from an Unvaccinated Horse in 2015. Viruses 2018, 10, 31. https://doi.org/10.3390/v10010031
Sreenivasan CC, Jandhyala SS, Luo S, Hause BM, Thomas M, Knudsen DEB, Leslie-Steen P, Clement T, Reedy SE, Chambers TM, et al. Phylogenetic Analysis and Characterization of a Sporadic Isolate of Equine Influenza A H3N8 from an Unvaccinated Horse in 2015. Viruses. 2018; 10(1):31. https://doi.org/10.3390/v10010031
Chicago/Turabian StyleSreenivasan, Chithra C., Sunayana S. Jandhyala, Sisi Luo, Ben M. Hause, Milton Thomas, David E. B. Knudsen, Pamela Leslie-Steen, Travis Clement, Stephanie E. Reedy, Thomas M. Chambers, and et al. 2018. "Phylogenetic Analysis and Characterization of a Sporadic Isolate of Equine Influenza A H3N8 from an Unvaccinated Horse in 2015" Viruses 10, no. 1: 31. https://doi.org/10.3390/v10010031
APA StyleSreenivasan, C. C., Jandhyala, S. S., Luo, S., Hause, B. M., Thomas, M., Knudsen, D. E. B., Leslie-Steen, P., Clement, T., Reedy, S. E., Chambers, T. M., Christopher-Hennings, J., Nelson, E., Wang, D., Kaushik, R. S., & Li, F. (2018). Phylogenetic Analysis and Characterization of a Sporadic Isolate of Equine Influenza A H3N8 from an Unvaccinated Horse in 2015. Viruses, 10(1), 31. https://doi.org/10.3390/v10010031