Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
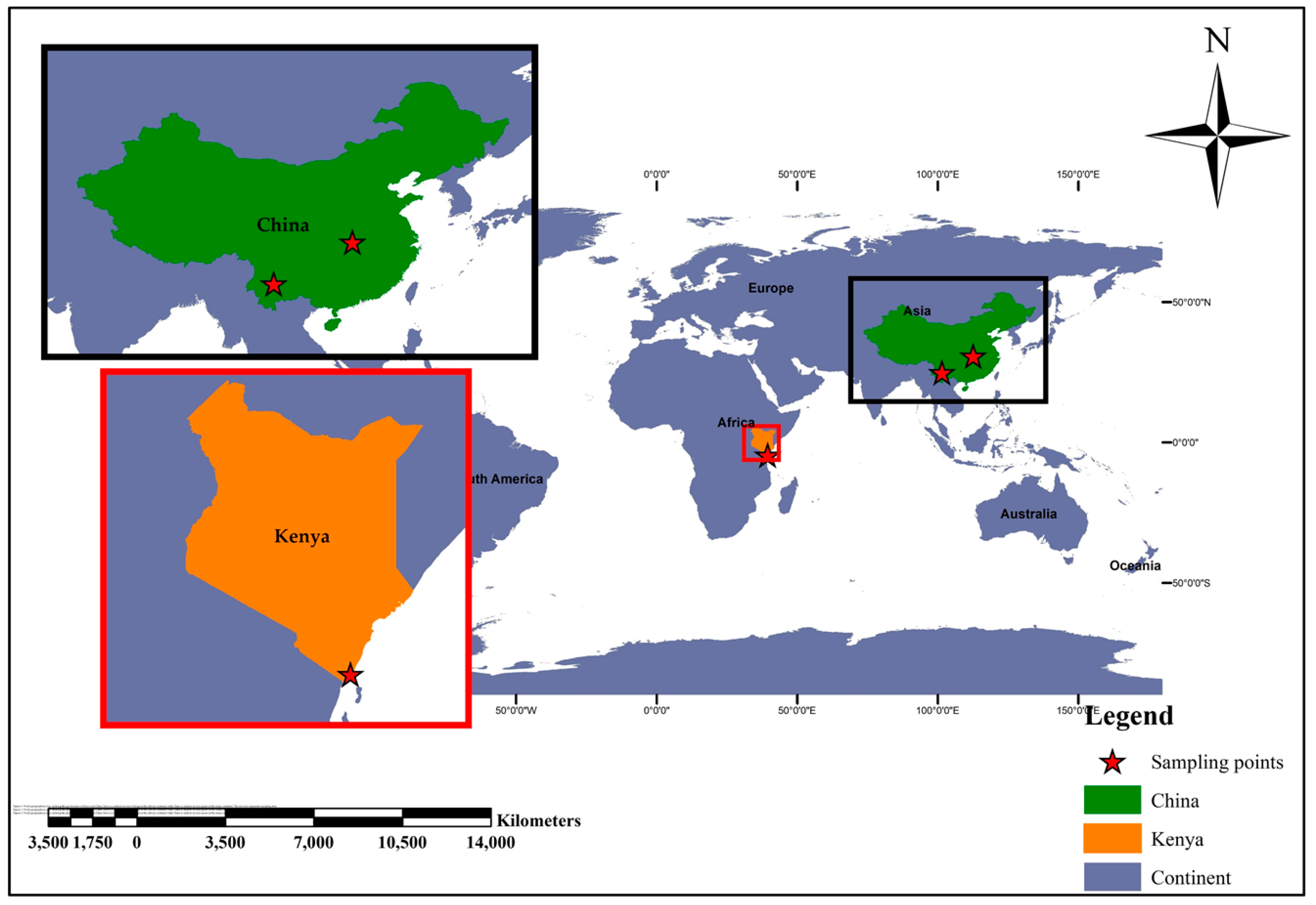
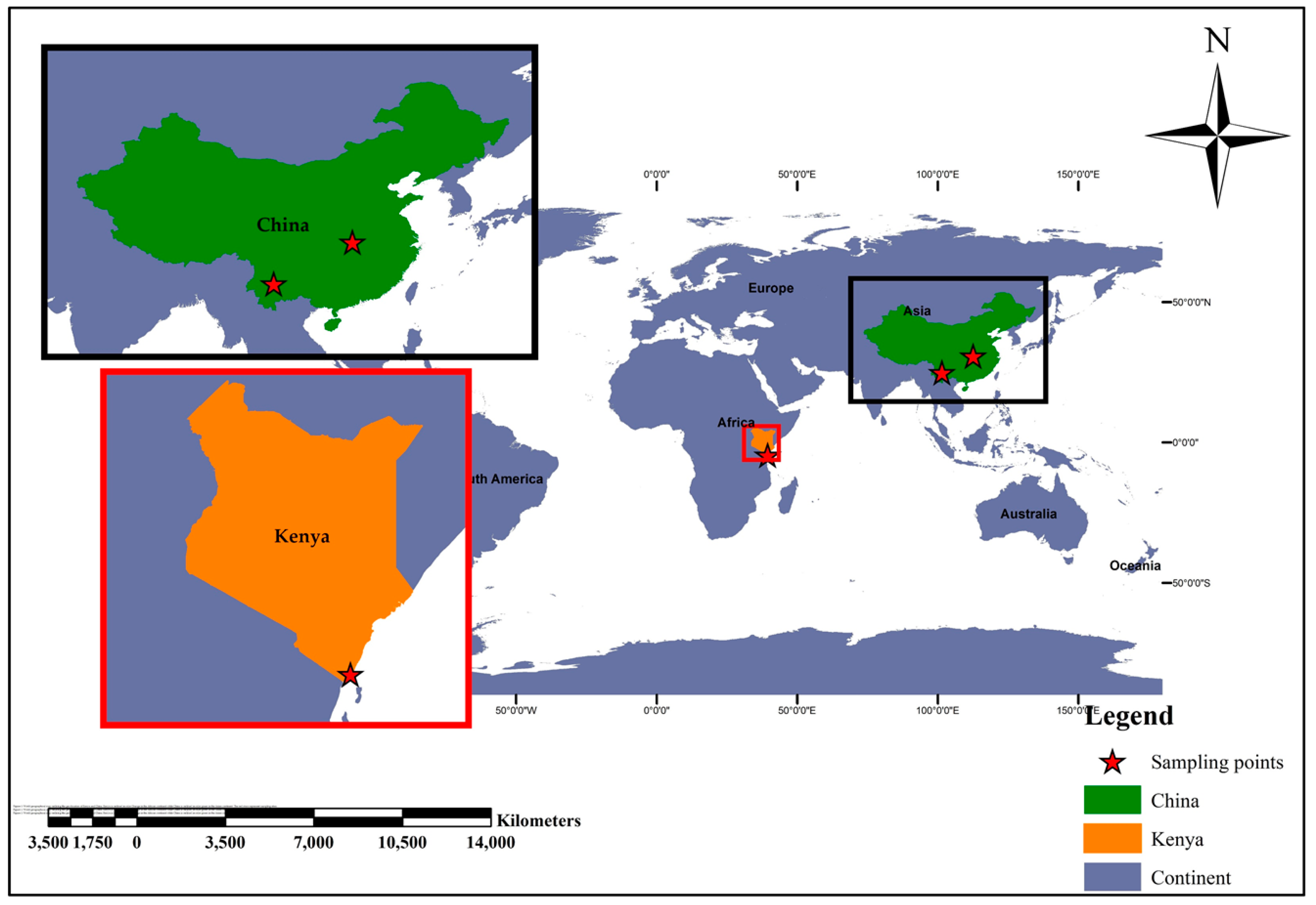
2.1. Study Sampling Sites and Related Important Ecological Features
2.1.1. Kwale County—Kenya
2.1.2. Kilifi County—Kenya
2.1.3. Hubei Province—China
2.1.4. Yunnan Province—China
2.2. Sample Collection and Taxonomic Identification
2.3. Viral RNA Isolation, cDNA Library Preparation, Amplification and Sequencing
2.4. Bioinformatics Analysis
2.5. Confirmation of Viral Sequences
3. Results
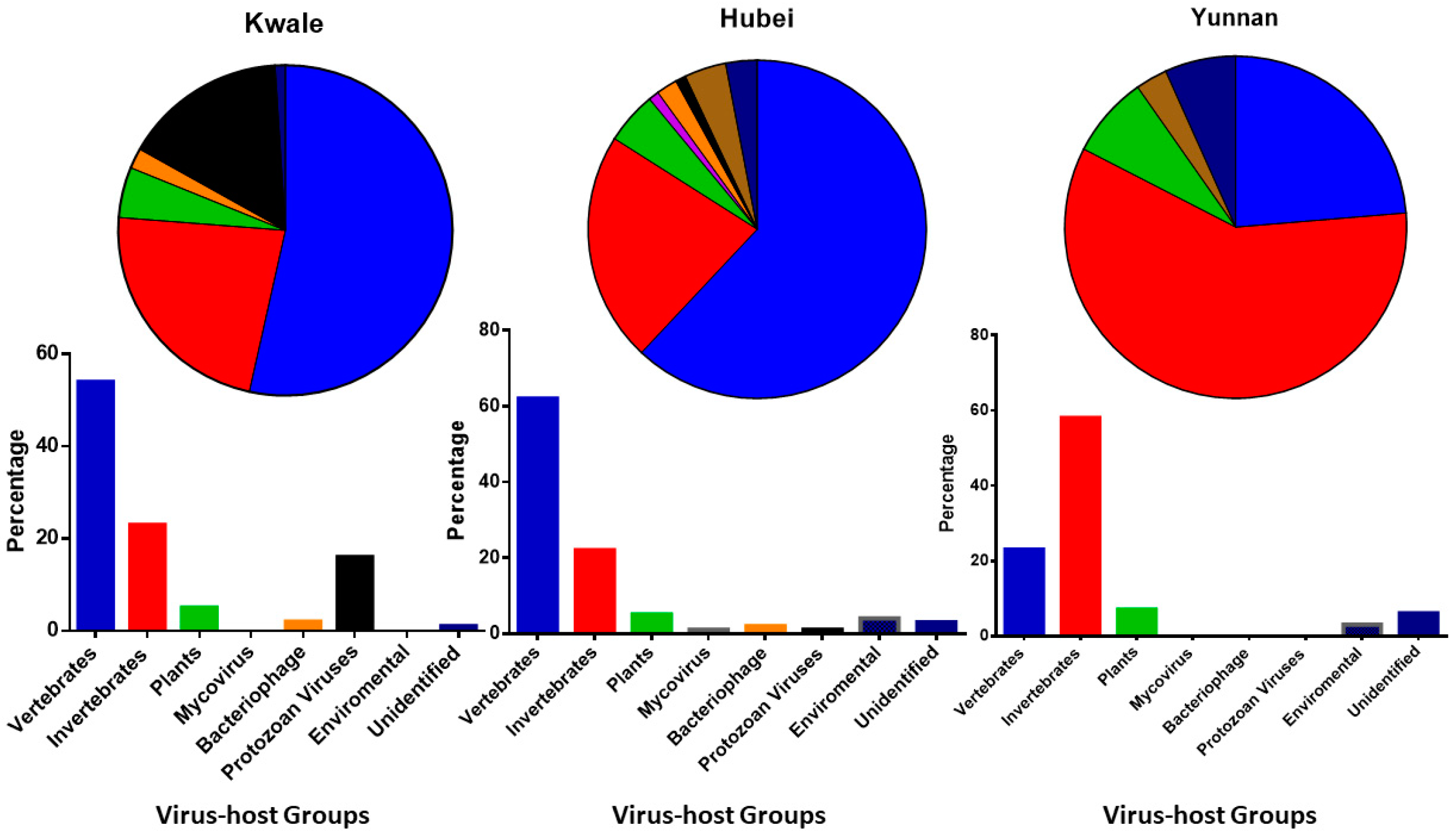
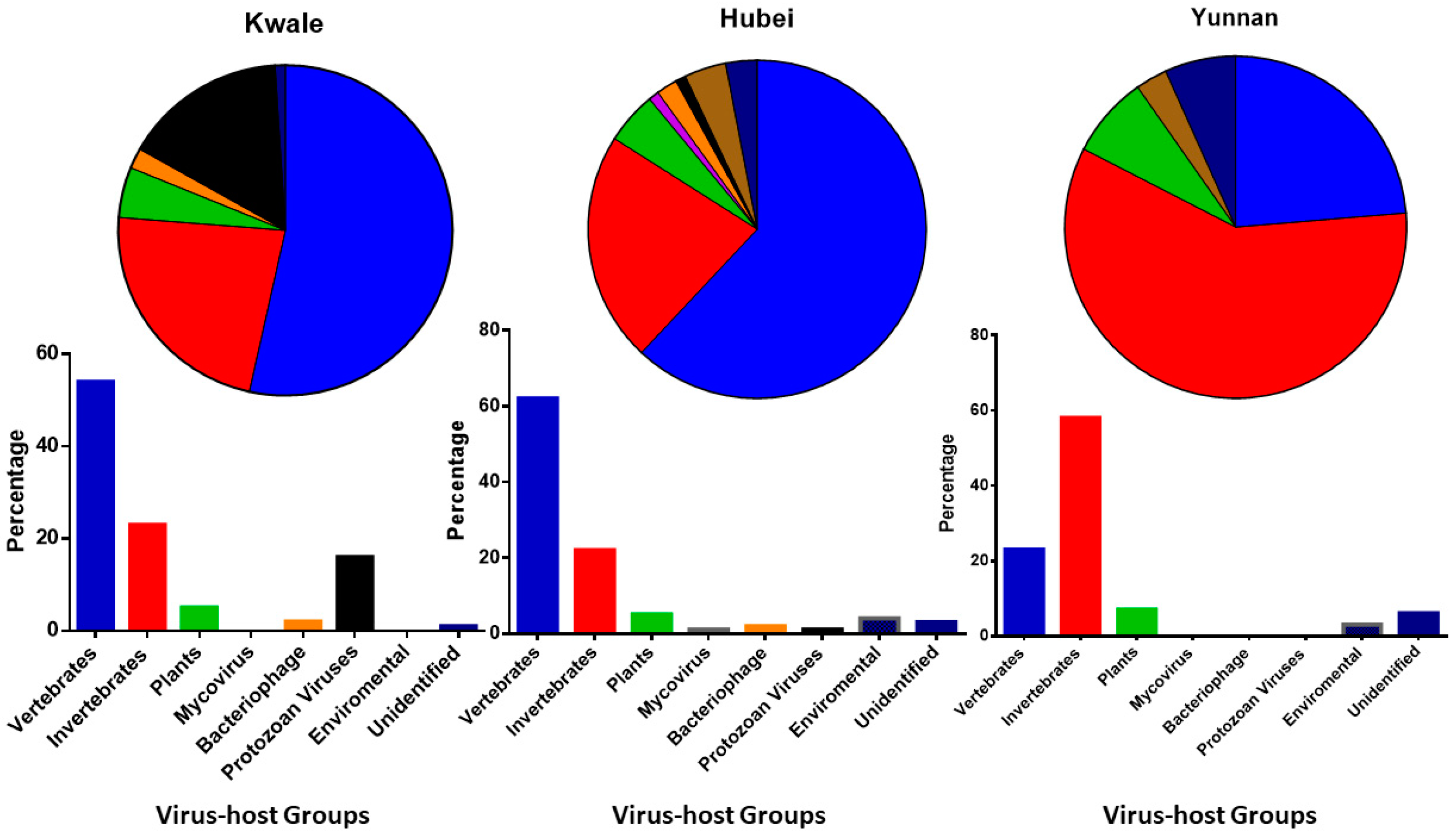
3.1. Proportion of Viruses in Host Groups
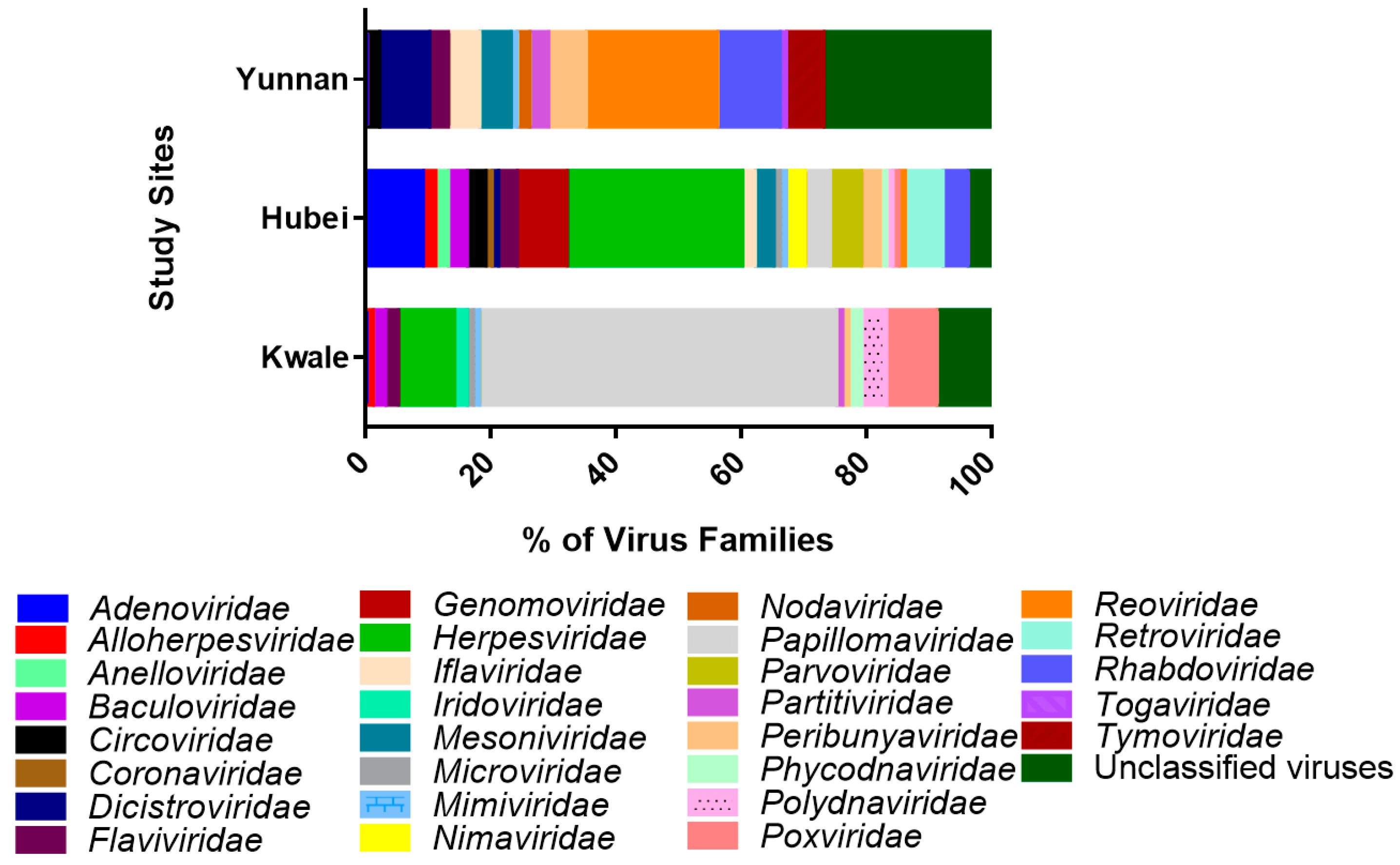
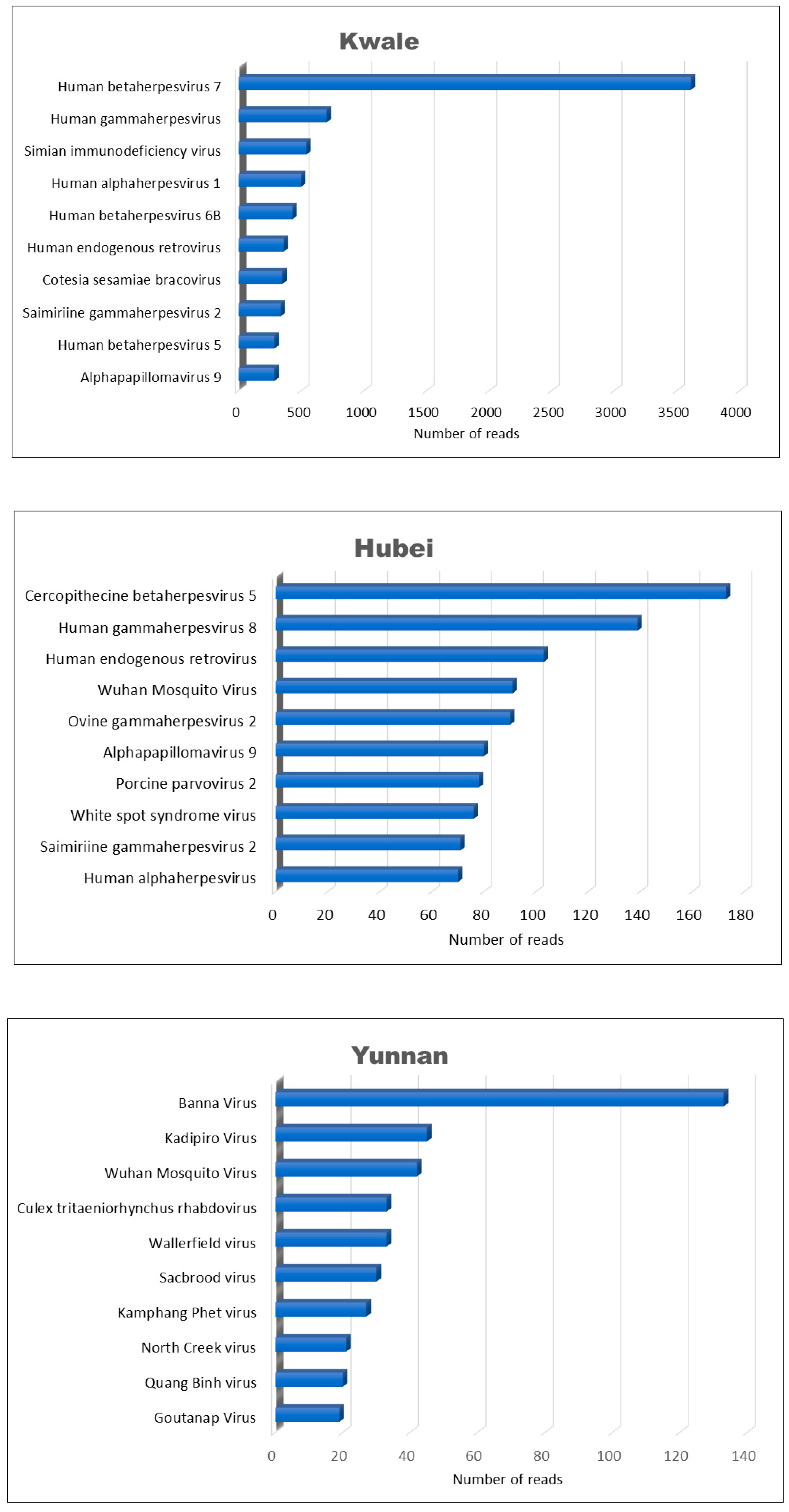
3.2. Virus Families
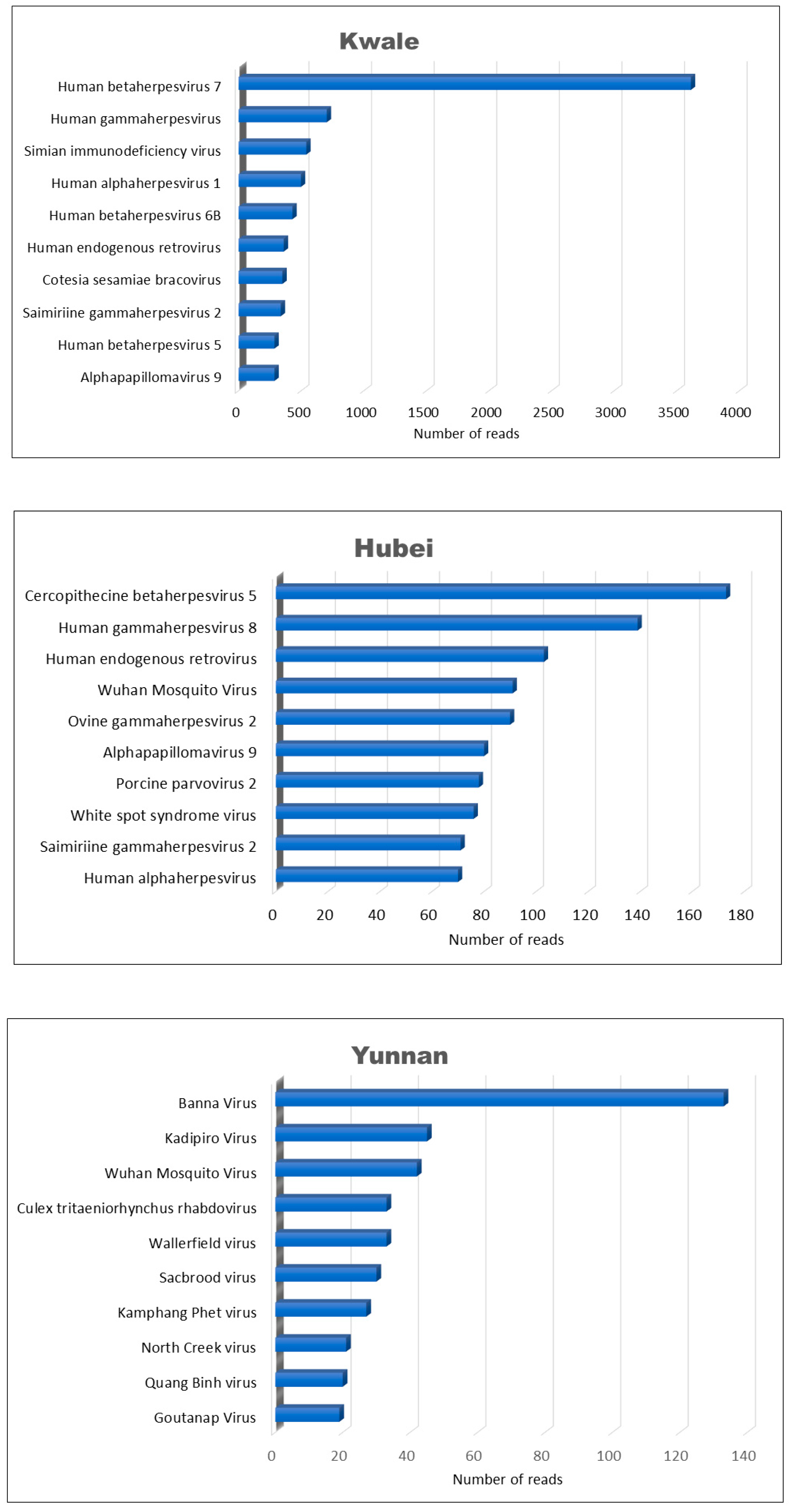
3.3. Viromes across the Three Locations
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brinkmann, A.; Nitsche, A.; Kohl, C. Viral Metagenomics on Blood-Feeding Arthropods as a Tool for Human Disease Surveillance. Int. J. Mol. Sci. 2016, 17, 1743. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; Diclaro, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles Gambiae Mosquitoes Harbor a Taxonomically Diverse Virome Including New Insect-Specific Flaviviruses, Mononegaviruses, and Totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Biser, T.; Hamilton, T.; Santos, C.J.; Pimentel, G.; Mokashi, V.P.; Bishop-Lilly, K.A. Bioinformatic Characterization of Mosquito Viromes within the Eastern United States and Puerto Rico: Discovery of Novel Viruses. Evol. Bioinform. 2016, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Liu, Y.; Hu, X.; Xiong, J.; Zhang, B.; Yuan, Z. A Metagenomic Survey of Viral Abundance and Diversity in Mosquitoes from Hubei Province. PLoS ONE 2015, 10, e0129845. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L. Viral Metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Moormann, A.M.; Snider, C.J.; Chelimo, K. The Company Malaria Keeps: How Co-Infection with Epstein-Barr Virus Leads to Endemic Burkitt Lymphoma. Curr. Opin. Infect. Dis. 2011, 24, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Sharma, S.; Krajacich, B.J.; Fakoli, L.S., III; Bolay, F.K.; Diclaro, J.W., II; Johnson, W.E.; Ebel, G.D.; Foy, B.D.; Brackney, D.E. Xenosurveillance: A Novel Mosquito-Based Approach for Examining the Human-Pathogen Landscape. PLoS Negl. Trop. Dis. 2015, 9, e0003628. [Google Scholar] [CrossRef] [PubMed]
- Stollar, V.; Thomas, V.L. An Agent in the Aedes Aegypti Cell Line (Peleg) Which Causes Fusion of Aedes albopictus Cells. Virology 1975, 64, 367–377. [Google Scholar] [CrossRef]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Olea-Popelka, F.J.; Eisen, L.; Moore, C.G.; Blair, C.D. Transmission Dynamics of an Insect-Specific Flavivirus in a Naturally Infected Culex pipiens Laboratory Colony and Effects of Co-Infection on Vector Competence for West Nile Virus. Virology 2012, 427, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Burivong, P.; Pattanakitsakul, S.-N.; Thongrungkiat, S.; Malasit, P.; Flegel, T.W. Markedly Reduced Severity of Dengue Virus Infection in Mosquito Cell Cultures Persistently Infected with Aedes albopictus Densovirus (AalDNV). Virology 2004, 329, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Karpf, A.R.; Lenches, E.; Strauss, E.G.; Strauss, J.H.; Brown, D.T. Superinfection Exclusion of Alphaviruses in Three Mosquito Cell Lines Persistently Infected with Sindbis Virus. J. Virol. 1997, 71, 7119–7123. [Google Scholar] [PubMed]
- Cholleti, H.; Hayer, J.; Abilio, A.P.; Mulandane, C.; Verner-carlsson, J.; Falk, K.I.; Fafetine, J.M.; Berg, M.; Blomström, A. Discovery of Novel Viruses in Mosquitoes from the Zambezi Valley of Mozambique. PLoS ONE 2016, 11, e0162751. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.J.; Hunt, R.H. Aedes albopictus in Africa? First Records of Live Specimens in Imported Tires in Cape Town. J. Am. Mosq. Control Assoc. 1991, 7, 107–108. [Google Scholar] [PubMed]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic Analysis of Viruses from Bat Fecal Samples Reveals Many Novel Viruses in Insectivorous Bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Li, Z.; Yang, F.; Zheng, J.; Feng, Y.; Guo, H.; Li, Y.; Wang, Y.; Su, N.; Zhang, F.; et al. Virome Profiling of Bats from Myanmar by Metagenomic Analysis of Tissue Samples Reveals More Novel Mammalian Viruses. PLoS ONE 2013, 8, e61950. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Sun, X.; Fu, S.; Wang, H.; Feng, Y.; Wang, H.; Tang, Q.; Liang, G.D. Distribution of Mosquitoes and Mosquito-Borne Arboviruses in Yunnan Province Near the China-Myanmar-Laos Border. Am. J. Trop. Med. Hyg. 2011, 84, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Pan, X.-L.; Zhang, H.-L.; Fu, S.-H.; Wang, H.-Y.; Tang, Q.; Wang, L.-F.; Liang, G.-D. Japanese Encephalitis Viruses from Bats in Yunnan, China. Emerg. Infect. Dis. 2009, 15, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Edwards, F. Mosquitoes of the Ethiopian Region III—Culicine Adults and Pupae | Mosquito Taxonomic Inventory; British Museum (Natural History): London, UK, 1941. [Google Scholar]
- Hopkins, G. Mosquitoes of the Ethiopian Region I. Larval Bionomics of Mosquitoes and Taxonomy of Culicine Larvae; British Museum (Natural History): London, UK, 1952. [Google Scholar]
- Carissimo, G.; van den Beek, M.; Vernick, K.D.; Antoniewski, C.; Schweer, T.; Yarza, P.; Schneider, D. Metavisitor, a Suite of Galaxy Tools for Simple and Rapid Detection and Discovery of Viruses in Deep Sequence Data. PLoS ONE 2017, 12, e0168397. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- GitHub. Taxonomizr. Available online: https://github.com/sherrillmix/taxonomizr (accessed on 23 August 2017).
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Molaei, G.; Andreadis, T.G.; Armstrong, P.M.; Dennett, J.A.; Real, S.V.; Sargent, C.; Bala, A.; Randle, Y.; Guzman, H.; Wuithiranyagool, T.; et al. Host Feeding Pattern of Culex quinquefasciatus (Diptera: Culicidae) and Its Role in Transmission of West Nile Virus in Harris Country, Texas. Annu. J. Trop. Med. Hyg. 2007, 77, 73–81. [Google Scholar]
- Burkett-Cadena, N.D.; Bingham, A.M.; Porterfield, C.; Unnasch, T.R. Innate Preference or Opportunism: Mosquitoes Feeding on Birds of Prey at the Southeastern Raptor Center. J. Vector Ecol. 2014, 39, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Börstler, J.; Jöst, H.; Garms, R.; Krüger, A.; Tannich, E.; Becker, N.; Schmidt-Chanasit, J.; Lühken, R. Host-Feeding Patterns of Mosquito Species in Germany. Parasit. Vectors 2016, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Sawabe, K.; Isawa, H.; Hoshino, K.; Sasaki, T.; Roychoudhury, S.; Higa, Y.; Kasai, S.; Tsuda, Y.; Nishiumi, I.; Hisai, N.; et al. Host-Feeding Habits of Culex pipiens and Aedes albopictus (Diptera: Culicidae) Collected at the Urban and Suburban Residential Areas of Japan. J. Med. Entomol. 2010, 47, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Takken, W.; Verhulst, N.O. Host Preferences of Blood-Feeding Mosquitoes. Annu. Rev. Entomol. 2013, 58, 433–453. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Willner, D.L.; Lim, Y.W.; Schmieder, R.; Chau, B.; Nilsson, C.; Anthony, S.; Ruan, Y.; Rohwer, F.; Breitbart, M. Broad Surveys of DNA Viral Diversity Obtained through Viral Metagenomics of Mosquitoes. PLoS ONE 2011, 6, e20579. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: The Classification and Nomenclature of Viruses the Online (10th) Report of the ICTV; ICTV: Budapest, Hungary, 2016. [Google Scholar]
- Attoui, H.; Mohd Jaafar, F.; Belhouchet, M.; Tao, S.; Chen, B.; Liang, G.; Tesh, R.B.; de Micco, P.; de Lamballerie, X. Liao Ning Virus, a New Chinese Seadornavirus That Replicates in Transformed and Embryonic Mammalian Cells. J. Gen. Virol. 2006, 87, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel Seadornavirus (Family Reoviridae) Related to Banna Virus in Europe. Arch. Virol. 2013, 158, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; He, Y.; Zhou, Y.; Meng, J.; Zhu, W.; Chen, H.; Liao, D.; Man, Y. Isolation and Genetic Characterization of Mangshi Virus: A Newly Discovered Seadornavirus of the Reoviridae Family Found in Yunnan Province, China. PLoS ONE 2015, 10, e0143601. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.P. First Isolation of New Orbivirus (BANNA) from Ticks and Infected Cattle Sera in Xinjiang. Endem. Dis. Bull. 1992, 7, 64–69. [Google Scholar]
- Xu, P.; Wang, Y.; Zuo, J.; Che, Y.; Peng, H.; Huang, Z.; Tang, G.; Xu, P.; Institute of Virology; et al. Recovery of the Same Type of Virus as Human New Orbivirus from Sera of Cattles and Pigs Collected in Yunnan Province. Chin. J. Virol. 1990, 6, 327–331. [Google Scholar]
- Xu, P.; Wang, Y.; Zuo, J.; Lin, J.; Xu, P. New Orbiviruses Isolated from Patients with Unknown Fever and Encephalitis in Yunnan Province. Chin. J. Virol. 1990, 6, 27–33. [Google Scholar]
- Tao, S.; Chen, B. Studies of Coltivirus in China. Chin. Med. J. (Engl.) 2005, 118, 581–586. [Google Scholar] [PubMed]
- Nabeshima, T.; Nga, P.T.; Guillermo, P.; Parquet, M.; Yu, F.; Thuy, N.T.; Trang, B.M.; Hien, N.T.; Nam, V.S.; Inoue, S.; et al. Isolation and Molecular Characterization of Banna Virus from Mosquitoes, Vietnam. Emerg. Infect. Dis. 2008, 14, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.E.; Gorman, B.M.; Tesh, R.B.; Knudson, D.L. Coltiviruses Isolated from Mosquitoes Collected in Indonesia. Virology 1993, 196, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fu, S.; Tian, Z.; He, Y.; Wang, H.; Wang, H.; Yang, H.; Tao, B.; Liang, G. Isolation and Identification of Banna Virus from Mosquito for the First Time in Inner Mongolia. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi 2009, 23, 106–108. [Google Scholar] [PubMed]
- Sun, X.; Meng, W.; Fu, S.; Feng, Y.; Zhai, Y.; Wang, J.; Wang, H.; Lv, X.; Liang, G. The First Report of Kadipiro Virus Isolation in China. Chin. J. Virol. 2009, 25, 173–177. [Google Scholar]
- Davison, A.J. Overview of Classification; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Roizman, B.; Carmichael, L.E.; Deinhardt, F.; de-The, G.; Nahmias, A.J.; Plowright, W.; Rapp, F.; Sheldrick, P.; Takahashi, M.; Wolf, K. Herpesviridae. Definition, Provisional Nomenclature, and Taxonomy. The Herpesvirus Study Group, the International Committee on Taxonomy of Viruses. Intervirology 1981, 16, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Mirandola, P.; Menotti, L. Human Herpesvirus 6: An Emerging Pathogen. Emerg. Infect. Dis. 1999, 5, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Elmore, D.; Eberle, R. Monkey B Virus (Cercopithecine Herpesvirus 1). Comp. Med. 2008, 58, 11–21. [Google Scholar] [PubMed]
- Takashima, Y.; Otsuka, H. Pathogenesis of Animal Herpesviruses to Human. Nihon Rinsho 2000, 58, 957–961. [Google Scholar] [PubMed]
- McHardy, J.; Williams, E.H.; Geser, A.; de-Thé, G.; Beth, E.; Giraldo, G. Endemic Kaposi’s Sarcoma: Incidence and Risk Factors in the West Nile District of Uganda. Int. J. Cancer 1984, 33, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, M.; Manno, D.; Guzzinati, S.; Tognazzo, S.; Zambon, P.; Arcà, B.; Costantini, C.; Ascoli, V. The Bloodsucking Arthropod Bite as Possible Cofactor in the Transmission of Human Herpesvirus-8 Infection and in the Expression of Kaposi’s Sarcoma Disease. Parassitologia 2002, 44, 123–129. [Google Scholar] [PubMed]
- Ascoli, V.; Manno, D.; Coluzzi, M. Geographic Variation in Human Herpesvirus 8 Seroprevalence and Possible Association with Exposure to Bites from Blood-Sucking Arthropods. J. Infect. Dis. 2006, 194, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Wodak, E.; Richter, S.; Bagó, Z.; Revilla-Fernández, S.; Weissenböck, H.; Nowotny, N.; Winter, P. Detection and Molecular Analysis of West Nile Virus Infections in Birds of Prey in the Eastern Part of Austria in 2008 and 2009. Vet. Microbiol. 2011, 149, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.; Tian, J.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, J.; Eden, J.; et al. Article Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Herpesvirus Systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Félez-Sánchez, M. Papillomaviruses: Viral Evolution, Cancer and Evolutionary Medicine. Evol. Med. Public Health 2015, 2015, 32–51. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.-M.; Fauquet, C.; Broker, T.R.; Bernard, H.-U.; zur Hausen, H. Classification of Papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.F.; Meijer, C.J.; International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic Classification of Human Papillomavirus Types Associated with Cervical Cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, A.; Forslund, O.; Ekberg, H.; Sterner, G.; Hansson, B.G. The Ubiquity and Impressive Genomic Diversity of Human Skin Papillomaviruses Suggest a Commensalic Nature of These Viruses. J. Virol. 2000, 74, 11636–11641. [Google Scholar] [CrossRef] [PubMed]
- Lutomiah, J.; Barrera, R.; Makio, A.; Mutisya, J.; Koka, H.; Owaka, S.; Koskei, E.; Nyunja, A.; Eyase, F.; Coldren, R.; et al. Dengue Outbreak in Mombasa City, Kenya, 2013–2014: Entomologic Investigations. PLoS Negl. Trop. Dis. 2016, 10, e0004981. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, C.N.; Price, M.A.; Fields, B.; Bonventure, J.; Ochieng, C.; Mwashigadi, G.; Hassan, A.S.; Thiong, A.N.; Micheni, M.; Mugo, P.; et al. Dengue and Chikungunya Virus Infections among Young Febrile Adults Evaluated for Acute HIV-1 Infection in Coastal Kenya. PLoS ONE 2016, 11, e0167508. [Google Scholar] [CrossRef] [PubMed]
- Sang, R.C. Dengue in Africa. J. Med. Entomol. 2005, 2, 3–6. [Google Scholar]
- Sun, X.; Fu, S.; Gong, Z.; Ge, J.; Meng, W.; Feng, Y.; Wang, J.; Zhai, Y.; Wang, H.; Nasci, R.; et al. Distribution of Arboviruses and Mosquitoes in Northwestern Yunnan Province, China. Vector-Borne Zoonotic Dis. 2009, 9, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, H.-J.; Liu, Z.-J.; Jing, J.; Ren, J.-Q.; Liu, Y.-Y.; Lu, F.; Jin, N.-Y. Japanese Encephalitis Virus in Mosquitoes and Swine in Yunnan Province, China 2009–2010. Vector-Borne Zoonotic Dis. 2013, 13, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, H.L.; Yang, W.H.; Zhang, Y.Z.; Huang, L.J.; Deng, S.Z.; Sun, Y.J.; Yang, D.J.; Zhou, J.H. Molecular Epidemiology of Japanese Encephalitis Viruses Isolated in Yunnan Province, 1977–2010. Zhonghua Liu Xing Bing Xue Za Zhi 2016, 37, 1519–1525. [Google Scholar] [PubMed]
- Lu, Z.; Fu, S.-H.; Cao, L.; Tang, C.-J.; Zhang, S.; Li, Z.-X.; Tusong, M.; Yao, X.-H.; Zhang, H.-L.; Wang, P.-Y.; et al. Human Infection with West Nile Virus, Xinjiang, China, 2011. Emerg. Infect. Dis. 2014, 20, 1421–1423. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Fu, S.-H.; Liu, W.-B.; Wang, H.-Y.; Lu, Z.; Tong, S.-X.; Li, Z.-X.; Nasci, R.S.; Kosoy, O.; Cui, Y.; et al. West Nile Virus Infection in Xinjiang, China. Vector-Borne Zoonotic Dis. 2013, 13, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Fu, S.; Lv, Z.; Tang, C.; Cui, S.; Li, X.; Gao, X.; Li, M.; Cao, Y.; Lei, W.; et al. West Nile Virus Infection in Suspected Febrile Typhoid Cases in Xinjiang, China. Emerg. Microbes Infect. 2017, 6, e41. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Nasci, R.S.; Godsey, M.S.; Savage, H.M.; Lutwama, J.J.; Lanciotti, R.S.; Peters, C.J. First Field Evidence for Natural Vertical Transmission of West Nile Virus in Culex Univittatus Complex Mosquitoes from Rift Valley Province, Kenya. Am. J. Trop. Med. Hyg. 2000, 62, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Nyamwaya, D.; Wang’ondu, V.; Amimo, J.; Michuki, G.; Ogugo, M.; Ontiri, E.; Sang, R.; Lindahl, J.; Grace, D.; Bett, B. Detection of West Nile Virus in Wild Birds in Tana River and Garissa Counties, Kenya. BMC Infect. Dis. 2016, 16, 696. [Google Scholar] [CrossRef] [PubMed]
- Lwande, O.W.; Venter, M.; Lutomiah, J.; Michuki, G.; Rumberia, C.; Gakuya, F.; Obanda, V.; Tigoi, C.; Odhiambo, C.; Nindo, F.; et al. Whole Genome Phylogenetic Investigation of a West Nile Virus Strain Isolated from a Tick Sampled from Livestock in North Eastern Kenya. Parasites Vectors 2014, 7, 542. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.H.; Yang, F.; Zhang, X.; Xu, X.; Kou, G.H.; Lo, C.F. Whispovirus. Curr. Top. Microbiol. Immunol. 2009, 328, 197–227. [Google Scholar] [PubMed]
- White, S.E.; Becnel, J.J.; Undeen, A.H.; Rotstein, M.J.; Moser, B.A.; Cockburn, A.; Fukuda, T. Epizootiology and Transmission of a Newly Discovered Baculovirus from the Mosquitoes Culex nigripalpus and C. quinquefasciatus. J. Gen. Virol. 2001, 82, 275–282. [Google Scholar] [CrossRef]
- Becnel, J.J. Prospects for the Mosquito Baculovirus Cuninpv as a Tool for Mosquito Control. J. Am. Mosq. Control Assoc. 2006, 22, 523–526. [Google Scholar] [CrossRef]
- Stephan, D.; Siddiqua, M.; Ta Hoang, A.; Engelmann, J.; Winter, S.; Maiss, E. Complete Nucleotide Sequence and Experimental Host Range of Okra Mosaic Virus. Virus Genes 2008, 36, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lv, X.; Zhai, Y.; Fu, S.; Wang, D.; Rayner, S.; Tang, Q.; Liang, G. Genomic Characterization of a Novel Virus of the Family Tymoviridae Isolated from Mosquitoes. PLoS ONE 2012, 7, e39845. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Location | Coordinates | Mosquito Species | No. of Sequenced Reads | Average Length of Reads |
|---|---|---|---|---|---|
| Kenya | Kwale | 4.46057° S, 39.47795° E | Culex quinquefasciatus | 21,747,508 | 150 bp |
| China | Hubei | 30.8843° N, 112.5923° E | Culex tritaeniorhynchus | 6,714,707 | 125 bp |
| Yunnan | 24.9756° N, 101.4848° E | Culex tritaeniorhynchus | 36,277,174 | 398 bp |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China. Viruses 2018, 10, 30. https://doi.org/10.3390/v10010030
Atoni E, Wang Y, Karungu S, Waruhiu C, Zohaib A, Obanda V, Agwanda B, Mutua M, Xia H, Yuan Z. Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China. Viruses. 2018; 10(1):30. https://doi.org/10.3390/v10010030
Chicago/Turabian StyleAtoni, Evans, Yujuan Wang, Samuel Karungu, Cecilia Waruhiu, Ali Zohaib, Vincent Obanda, Bernard Agwanda, Morris Mutua, Han Xia, and Zhiming Yuan. 2018. "Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China" Viruses 10, no. 1: 30. https://doi.org/10.3390/v10010030
APA StyleAtoni, E., Wang, Y., Karungu, S., Waruhiu, C., Zohaib, A., Obanda, V., Agwanda, B., Mutua, M., Xia, H., & Yuan, Z. (2018). Metagenomic Virome Analysis of Culex Mosquitoes from Kenya and China. Viruses, 10(1), 30. https://doi.org/10.3390/v10010030