Therapeutic Approaches Using Host Defence Peptides to Tackle Herpes Virus Infections


Abstract
:1. Introduction
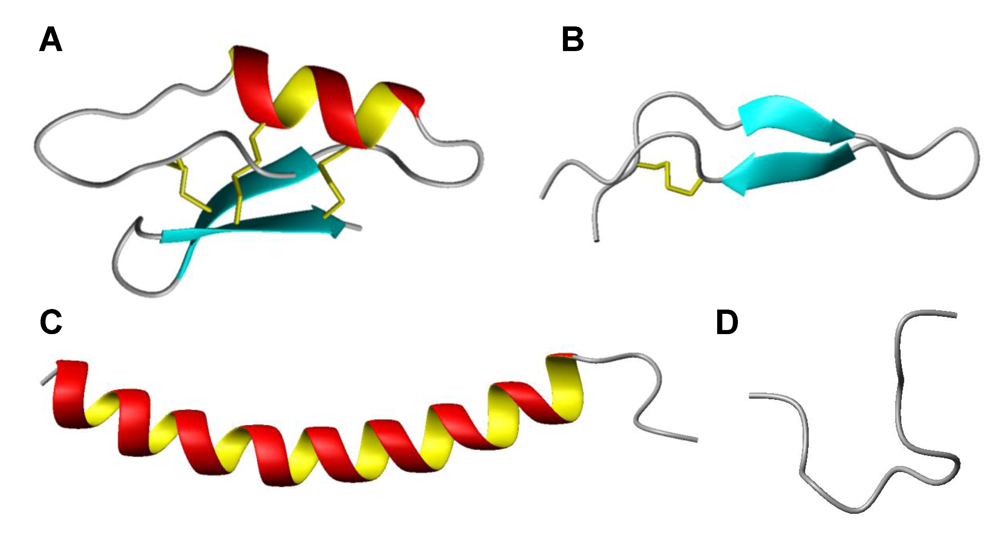
3. Host defence peptides
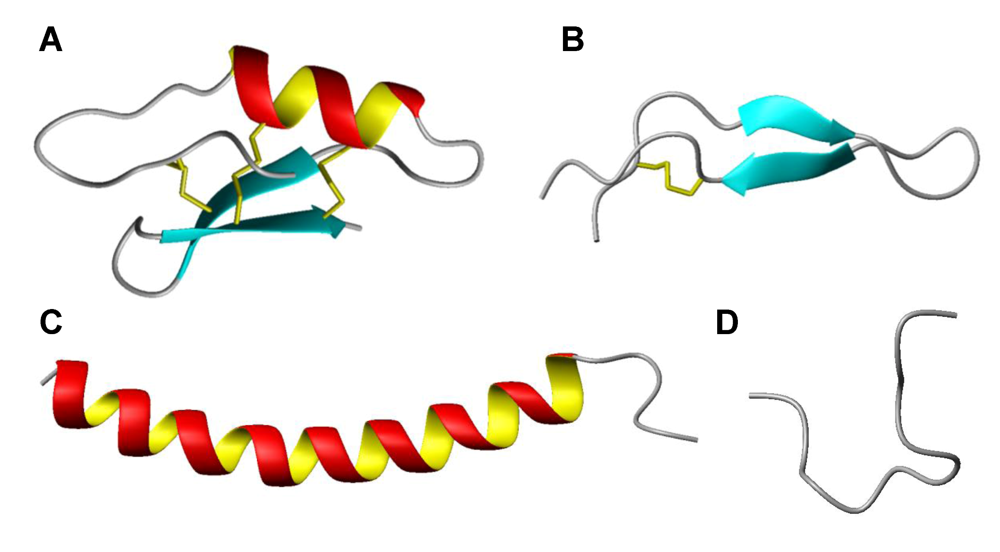
4. Antiviral activity of HDPs
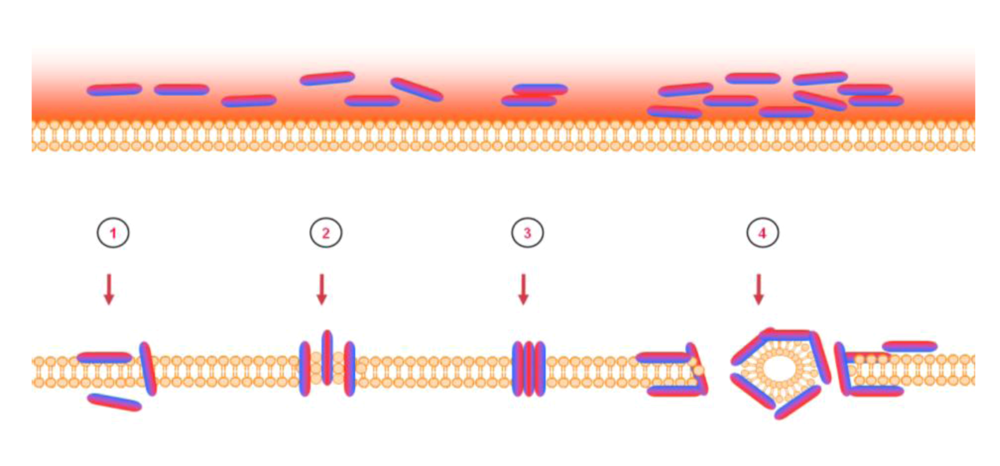
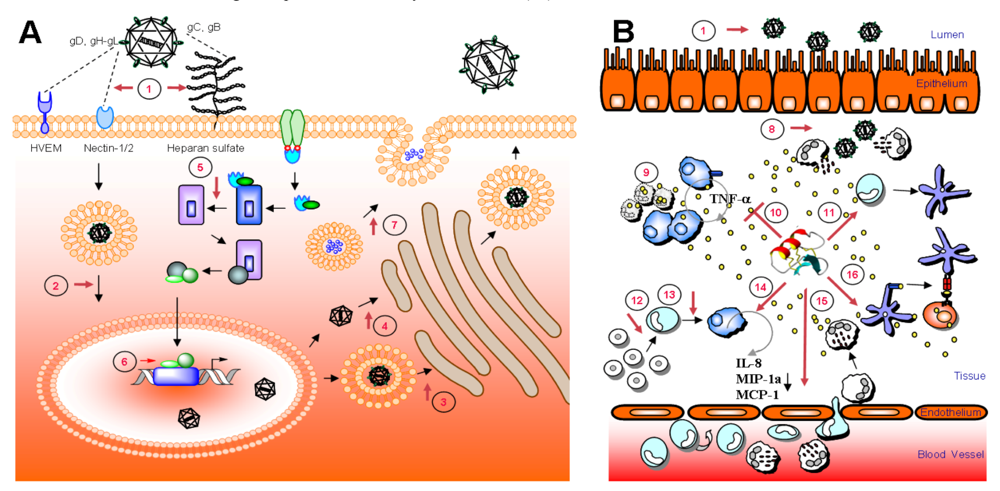
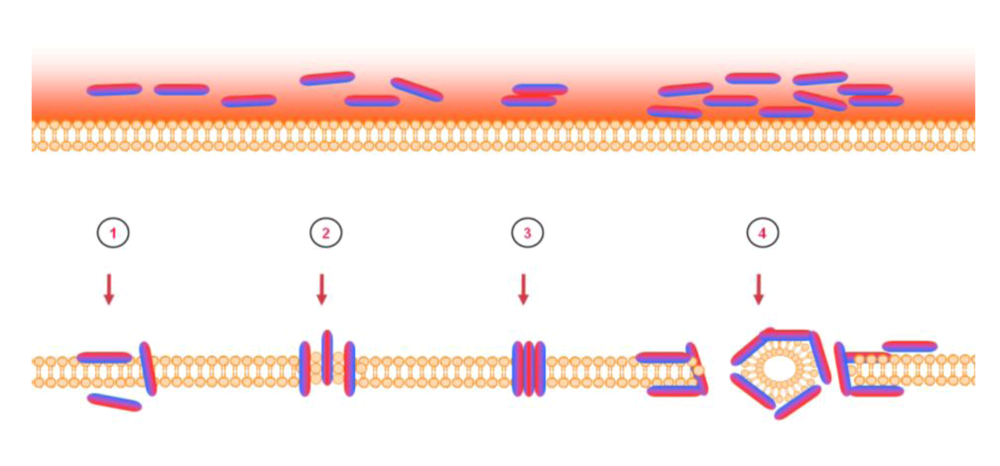
6. Mode of action – blocking of viral entry by heparan sulfate interaction

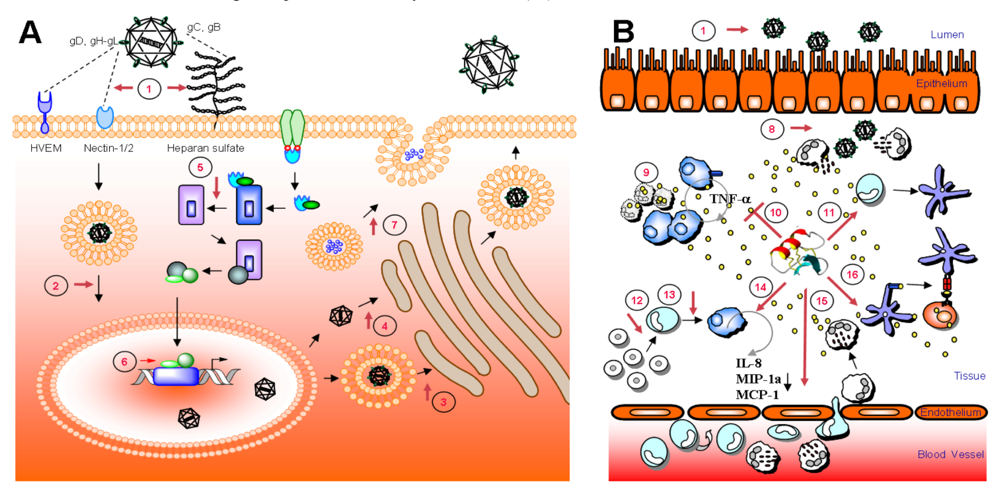{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Structure | Source | Virus | Primary amino acid sequence | Proposed antiviral mechanism | References |
|---|---|---|---|---|---|---|
| Magainin | α-helix | Frog | HSV | GIGKFLHSAKKFGKAFVGEIMNS | Cellular target | [56,57,71] |
| Cecropin A1 | α-helix | Insect | HSV | GWLKKIGKKIERVGQHTRDATIQGLGVAQQAANVAATAR | Cellular target | [57,72] |
| Melittin | α-helix | Bee | HSV | GIGAVLKVLTTGLPALISWIKRKRQQ | Cellular target | [55,57,73] |
| LL-37 | α-helix | Human | HSV | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | Weak viral inactivation | [55,74] |
| Brevinin-1 | α-helix | Frog | HSV | FLPVLAGIAAKVVPALFC1KITKKC1 | Viral inactivation | [55,75] |
| θ defensin | Cyclic β-sheet | Primate / human | HSV | G1FC2RC3LC4RRGVC4RC3IC2TR1 | Binds gB and blocks viral attachment | [64,76] |
| Defensin | β-sheet | Human / rabbit | HSVHCMV | MPC1SC2KKYC3DPWEVIDGSC2GLFNSKYIC3C1REK | Interacts with HSV membrane/ glycoprotein and cellular target but not heparan sulfateInactivates viral particle | [60,63,64,77,78] |
| Dermaseptin | β-sheet | Frog | HSV | ALWKTMLKKLGTMALHAGKAALGAAADTISQGTQ | Activity at virus-cell interface | [58,79] |
| Tachyplesin | β-sheet | Horse shoe crab | HSV | KWC1FRVC2YRGIC2YRRC1R | Viral inactivation | [55,80] |
| Protegrin | β-sheet | Human / porcine | HSV | RGGRLC1YC2RRRFC2VC1VGR | Viral inactivation | [55,81] |
| Lactoferricin | β-turn | Human / bovine | HSVHCMV | FKC1RRWQWRMKKLGAPSITC1VRRAFA | Blocks heparan sulfate, but a secondary effect has also been indicated.Activity at virus-cell interface | [59,61,82-85] |
| Indolicidin | Extended | Bovine | HSV | ILPWKWPWWPWRR | Targets viral membrane / glycoprotein | [47,57] |
8. Intracellular targets and host cell stimulation
9. Adjuvant potential of HDP derived peptides
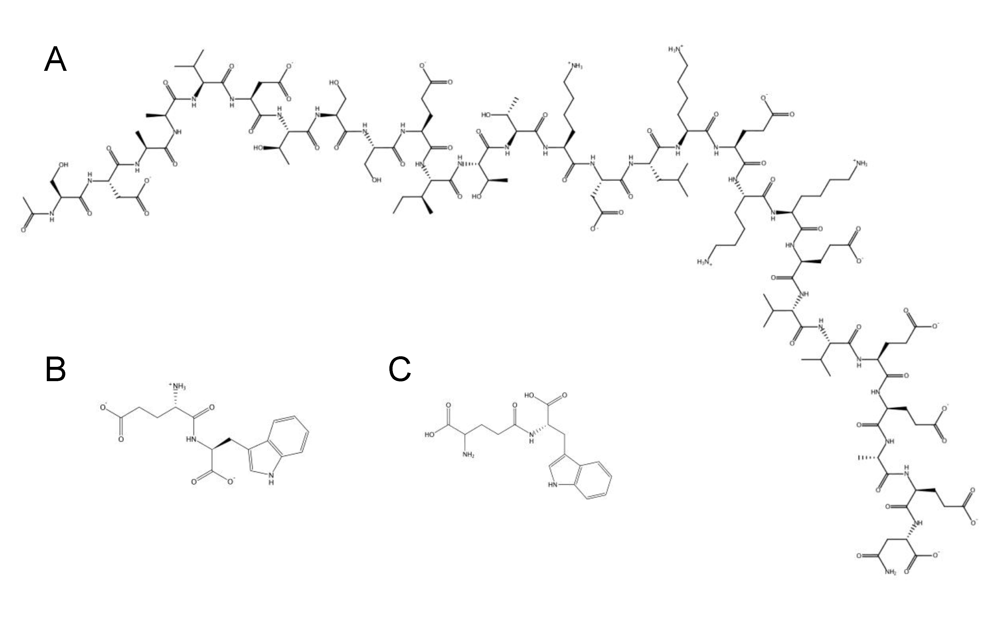
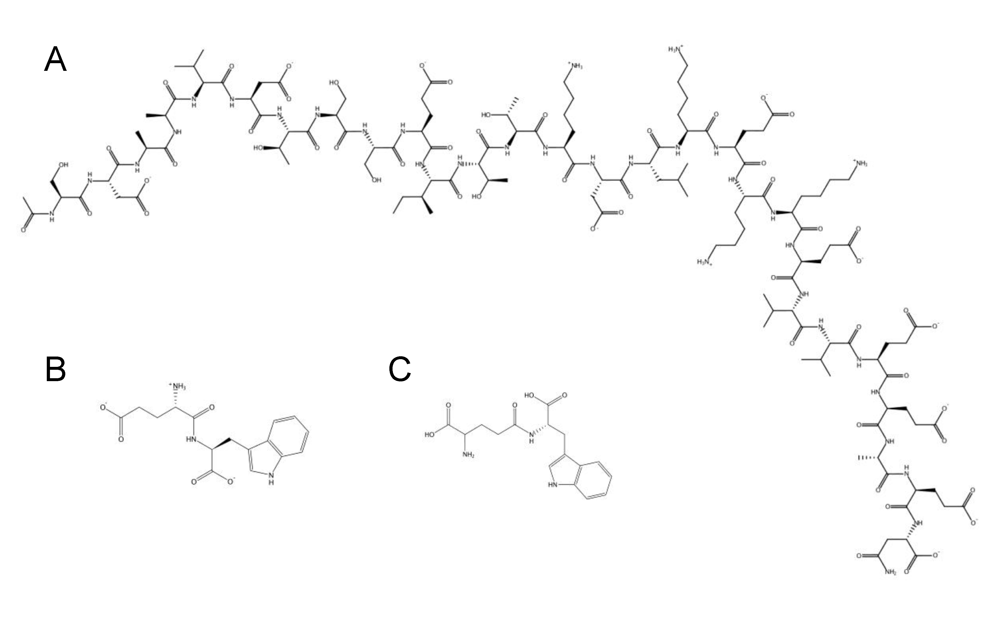
10. Clinical potential of HDPs as antiviral therapeutics
| Drug | Sequence | Description/Status/Results | Company & reference |
|---|---|---|---|
| Plectasin NZ2114 | GFGC1NGPWDEDDMQC2HNHC3KSIKGYKGGYC1AKGGFVC2KC3Y-COOH (Plectasin) | Preclinical: A variant of plectasin which has demonstrated potent Gram-positive effect in systemic pneumococcal and streptococcal infections. | Novozymes AS / Sanofi-Aventis (Bagsvœrd, Denmark), www.novozymes.com |
| Zadaxin℘/Thymosin α1/thymalfasin | AC-SDAAVDTSSEITTKDLKEKKEVVEEAEN-COOH | Phase III / approved: A 28-mer bovine HDP intended for directed therapy targeting chronic hepatitis B and C viruses. It promotes MHC class I expression, interleukin-2 and interferon-γ secretion, proliferation and activation of CD4 Th1, CD8, and NK cells. It also decreases Th2 cytokines (e.g., interleukin-4 and interleukin-10) that are counter productive to viral infections. | SciClone / Sigma-Tau (Foster City, CA, USA), www.hcvadvocate.org,www.scicloneinternational.com |
| Oglufanide disodium/IM862 | EW-COOH | Phase II: A di-peptide for direct targeting chronic hepatitis C virus. Administration of an intranasal synthetic formulation of IM862 has demonstrated to reverses the suppression of the immune system. | Implicit Bioscience (Toowong, QLD, Australia), www.hcvadvocate.org |
| SCV-07 | γ-D-glutamyl-L-tryptophan | Phase IIa: SCV-07 has demonstrated promising results as a mono-therapy in patients with chronic hepatitis C virus infection. | SciClonewww.scicloneinternational.com |
11. Conclusion
References
- www.who.int. World Health Statistics . Available online: www.who.int/whosis/whostat/2008/en/index.html (accessed 3 March 2008).
- Hamill, P.; Brown, K.; Jenssen, H.; Hancock, R.E.W. Novel anti-infectives: is host defence the answer? Curr. Opin. Biotechnol. 2008, 19, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gallo, R.L. Toll-like receptors in skin infections and inflammatory diseases. Infect. Disord. Drug Targets 2008, 8, 144–155. [Google Scholar] [PubMed]
- O'Neill, L.A. Targeting signal transduction as a strategy to treat inflammatory diseases. Nat. Rev. Drug Discov. 2006, 5, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Romagne, F. Current and future drugs targeting one class of innate immunity receptors: the Toll-like receptors. Drug Discov. Today 2007, 12, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Wales, J.; Andreakos, E.; Feldmann, M.; Foxwell, B. Targeting intracellular mediators of pattern-recognition receptor signalling to adjuvant vaccination. Biochem. Soc. Trans. 2007, 35, 1501–1503. [Google Scholar] [CrossRef] [PubMed]
- Esmann, J. The many challenges of facial herpes simplex virus infection. J. Antimicrob. Chemother. 2001, 47, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.M.; Bloom, D.C.; Cohrs, R.J.; Gilden, D.H.; Kennedy, P.G. Herpes simplex virus-1 and varicella-zoster virus latency in ganglia. J. Neurovirol. 2003, 9, 194–204. [Google Scholar] [PubMed]
- Shieh, M.T.; WuDunn, D.; Montgomery, R.I.; Esko, J.D.; Spear, P.G. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 1992, 116, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [PubMed]
- Mardberg, K.; Trybala, E.; Tufaro, F.; Bergstrom, T. Herpes simplex virus type 1 glycoprotein C is necessary for efficient infection of chondroitin sulfate-expressing gro2C cells. J. Gen. Virol. 2002, 83, 291–300. [Google Scholar] [PubMed]
- Gruenheid, S.; Gatzke, L.; Meadows, H.; Tufaro, F. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J. Virol. 1993, 67, 93–100. [Google Scholar] [PubMed]
- Banfield, B.W.; Leduc, Y.; Esford, L.; Visalli, R.J.; Brandt, C.R.; Tufaro, F. Evidence for an interaction of herpes simplex virus with chondroitin sulfate proteoglycans during infection. Virology 1995, 208, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.S.; Geraghty, R.J.; Martinez, W.M.; Montgomery, R.I.; Whitbeck, J.C.; Xu, R.; Eisenberg, R.J.; Cohen, G.H.; Spear, P.G. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 1998, 246, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar] [PubMed]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Chen, J.; Tiwari, V.; Ju, W.; Li, J.P.; Malmstrom, A.; Shukla, D.; Liu, J. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 2002, 277, 37912–37919. [Google Scholar] [CrossRef] [PubMed]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar] [PubMed]
- Balan, P.; Davis-Poynter, N.; Bell, S.; Atkinson, H.; Browne, H.; Minson, T. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 1994, 75, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Dingwell, K.S.; Johnson, D.C. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 1998, 72, 8933–8942. [Google Scholar] [PubMed]
- Creagh, E.M.; O'Neill, L.A. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Gupta, D. The peptidoglycan recognition proteins (PGRPs). Genome Biol. 2006, 7, 232. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, G.R.; Woolley, P.D.; Thin, R.N.; De Maubeuge, J.; Foidart, J.M.; Engst, R. Acyclovir vs isoprinosine (immunovir) for suppression of recurrent genital herpes simplex infection. Genitourin Med. 1992, 68, 312–316. [Google Scholar] [PubMed]
- Chilukuri, S.; Rosen, T. Management of acyclovir-resistant herpes simplex virus. Dermatol. Clin. 2003, 21, 311–320. [Google Scholar] [PubMed]
- De Clercq, E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004, 2, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hancock, R.E.W.; Cherkasov, A. AMPer: a database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E.W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.H.; Jenssen, H.; Sandvik, K.; Gutteberg, T.J. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med. Virol. 2004, 74, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Haukland, H.H.; Ulvatne, H.; Sandvik, K.; Vorland, L.H. The antimicrobial peptides lactoferricin B and magainin 2 cross over the bacterial cytoplasmic membrane and reside in the cytoplasm. FEBS Lett. 2001, 508, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lau, Y.E.; Rozek, A.; Scott, M.G.; Goosney, D.L.; Davidson, D.J.; Hancock, R.E.W. Interaction and cellular localization of the human host defense peptide LL-37 with lung epithelial cells. Infect. Immun. 2005, 73, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Speert, D.P.; Hancock, R.E.W. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J. Immunol. 2004, 172, 3758–3765. [Google Scholar] [PubMed]
- Wachinger, M.; Kleinschmidt, A.; Winder, D.; von Pechmann, N.; Ludvigsen, A.; Neumann, M.; Holle, R.; Salmons, B.; Erfle, V.; Brack-Werner, R. Antimicrobial peptides melittin and cecropin inhibit replication of human immunodeficiency virus 1 by suppressing viral gene expression. J. Gen. Virol. 1998, 79, 731–740. [Google Scholar] [PubMed]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sonksen, C.P.; Ludvigsen, S.; Raventos, D.; Buskov, S.; Christensen, B.; De Maria, L.; Taboureau, O.; Yaver, D.; Elvig-Jorgensen, S.G.; Sorensen, M.V.; Christensen, B.E.; Kjaerulff, S.; Frimodt-Moller, N.; Lehrer, R.I.; Zasloff, M.; Kristensen, H.H. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Hwang, P.M.; Zhou, N.; Shan, X.; Arrowsmith, C.H.; Vogel, H.J. Three-dimensional solution structure of lactoferricin B, an antimicrobial peptide derived from bovine lactoferrin. Biochemistry 1998, 37, 4288–4298. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of Human Host Defense Cathelicidin LL-37 and Its Smallest Antimicrobial Peptide KR-12 in Lipid Micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E.W. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51-55,29-32. [Google Scholar] [CrossRef]
- Bastian, A.; Schafer, H. Human alpha-defensin 1 (HNP-1) inhibits adenoviral infection in vitro. Regul. Pept. 2001, 101, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Wiethoff, C.M.; Cui, C.; Wilcoxen, K.M.; Amorin, M.; Ghadiri, M.R.; Nemerow, G.R. Antiviral cyclic D,L-alpha-peptides: targeting a general biochemical pathway in virus infections. Bioorg. Med. Chem. 2005, 13, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.B.; Lee, A.; Wan, J.; Roginski, H.; Coventry, M.J. The effect of bovine lactoferrin and lactoferricin B on the ability of feline calicivirus (a norovirus surrogate) and poliovirus to infect cell cultures. J. Appl. Microbiol. 2003, 95, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Pietrantoni, A.; Ammendolia, M.G.; Tinari, A.; Siciliano, R.; Valenti, P.; Superti, F. Bovine lactoferrin peptidic fragments involved in inhibition of Echovirus 6 in vitro infection. Antiviral Res. 2006, 69, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Benincasa, M.; Skerlavaj, B.; Gennaro, R.; Pellegrini, A.; Zanetti, M. In vitro and in vivo antimicrobial activity of two alpha-helical cathelicidin peptides and of their synthetic analogs. Peptides 2003, 24, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Ourth, D.D.; Lockey, T.D.; Renis, H.E. Induction of cecropin-like and attacin-like antibacterial but not antiviral activity in Heliothis virescens larvae. Biochem. Biophys. Res. Commun. 1994, 200, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Aboudy, Y.; Mendelson, E.; Shalit, I.; Bessalle, R.; Fridkin, M. Activity of two synthetic amphiphilic peptides and magainin-2 against herpes simplex virus types 1 and 2. Int. J. Pept. Protein Res. 1994, 43, 573–582. [Google Scholar] [PubMed]
- Albiol Matanic, V.C.; Castilla, V. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int. J. Antimicrob. Agents 2004, 23, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Belaid, A.; Aouni, M.; Khelifa, R.; Trabelsi, A.; Jemmali, M.; Hani, K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J. Med. Virol. 2002, 66, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.H.; Jenssen, H.; Gutteberg, T.J. Lactoferrin and lactoferricin inhibit Herpes simplex 1 and 2 infection and exhibit synergy when combined with acyclovir. Antiviral Res. 2003, 58, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar] [PubMed]
- Jenssen, H.; Andersen, J.H.; Uhlin-Hansen, L.; Gutteberg, T.J.; Rekdal, O. Anti-HSV activity of lactoferricin analogues is only partly related to their affinity for heparan sulfate. Antiviral Res. 2004, 61, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Szklarek, D.; Ganz, T.; Selsted, M.E. Correlation of binding of rabbit granulocyte peptides to Candida albicans with candidacidal activity. Infect. Immun. 1985, 49, 207–211. [Google Scholar] [PubMed]
- Sinha, S.; Cheshenko, N.; Lehrer, R.I.; Herold, B.C. NP-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrob. Agents Chemother. 2003, 47, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Yasin, B.; Wang, W.; Pang, M.; Cheshenko, N.; Hong, T.; Waring, A.J.; Herold, B.C.; Wagar, E.A.; Lehrer, R.I. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 2004, 78, 5147–5156. [Google Scholar] [CrossRef] [PubMed]
- Robinson Jr., W.E.; McDougall, B.; Tran, D.; Selsted, M.E. Anti-HIV-1 activity of indolicidin, an antimicrobial peptide from neutrophils. J. Leukoc. Biol. 1998, 63, 94–100. [Google Scholar] [PubMed]
- Jenssen, H.; Andersen, J.H.; Mantzilas, D.; Gutteberg, T.J. A wide range of medium-sized, highly cationic, alpha-helical peptides show antiviral activity against herpes simplex virus. Antiviral Res. 2004, 64, 119–126. [Google Scholar] [PubMed]
- Tamamura, H.; Murakami, T.; Masuda, M.; Otaka, A.; Takada, W.; Ibuka, T.; Nakashima, H.; Waki, M.; Matsumoto, A.; Yamamoto, N. Structure-activity relationships of an anti-HIV peptide, T22. Biochem. Biophys. Res. Commun. 1994, 205, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.G.; Hahm, K.S.; Shin, S.Y. Structure and fungicidal activity of a synthetic antimicrobial peptide, P18, and its truncated peptides. Biotechnol. Lett. 2004, 26, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Gutteberg, T.J.; Lejon, T. Modelling of anti-HSV activity of lactoferricin analogues using amino acid descriptors. J. Pept. Sci. 2005, 11, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, F.; Massucci, M.T.; Giardi, M.F.; Nozza, F.; Pulsinelli, E.; Nicolini, C.; Botti, D.; Antonini, G. Antiviral activity of ovotransferrin derived peptides. Biochem. Biophys. Res. Commun. 2005, 331, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Gesell, J.; Zasloff, M.; Opella, S.J. Two-dimensional 1H NMR experiments show that the 23-residue magainin antibiotic peptide is an alpha-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 1997, 9, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Onsins, S.; Aguade, M. Molecular evolution of the Cecropin multigene family in Drosophilafunctional genes vs. pseudogenes. Genetics 1998, 150, 157–171. [Google Scholar] [PubMed]
- Habermann, E.; Jentsch, J. [Sequence analysis of melittin from tryptic and peptic degradation products]. Hoppe Seylers Z Physiol. Chem. 1967, 348, 37–50. [Google Scholar] [PubMed]
- Gudmundsson, G.H.; Agerberth, B.; Odeberg, J.; Bergman, T.; Olsson, B.; Salcedo, R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur. J. Biochem. 1996, 238, 325–332. [Google Scholar] [PubMed]
- Conlon, J.M.; Al-Ghaferi, N.; Abraham, B.; Jiansheng, H.; Cosette, P.; Leprince, J.; Jouenne, T.; Vaudry, H. Antimicrobial peptides from diverse families isolated from the skin of the Asian frog, Rana grahami. Peptides 2006, 27, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Yuan, J.; Osapay, G.; Osapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Sawai, M.V.; Jia, H.P.; Liu, L.; Aseyev, V.; Wiencek, J.M.; McCray Jr., P.B.; Ganz, T.; Kearney, W.R.; Tack, B.F. The NMR structure of human beta-defensin-2 reveals a novel alpha-helical segment. Biochemistry 2001, 40, 3810–3816. [Google Scholar] [CrossRef] [PubMed]
- McManus, A.M.; Dawson, N.F.; Wade, J.D.; Carrington, L.E.; Winzor, D.J.; Craik, D.J. Three-dimensional structure of RK-1: a novel alpha-defensin peptide. Biochemistry 2000, 39, 15757–15764. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Nguyen, V.H.; Delfour, A.; Migliore-Samour, D.; Nicolas, P. Isolation, amino acid sequence, and synthesis of dermaseptin, a novel antimicrobial peptide of amphibian skin. Biochemistry 1991, 30, 8824–8830. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar] [PubMed]
- Aumelas, A.; Mangoni, M.; Roumestand, C.; Chiche, L.; Despaux, E.; Grassy, G.; Calas, B.; Chavanieu, A. Synthesis and solution structure of the antimicrobial peptide protegrin-1. Eur. J. Biochem. 1996, 237, 575–583. [Google Scholar] [PubMed]
- Andersen, J.H.; Osbakk, S.A.; Vorland, L.H.; Traavik, T.; Gutteberg, T.J. Lactoferrin and cyclic lactoferricin inhibit the entry of human cytomegalovirus into human fibroblasts. Antiviral Res. 2001, 51, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; Jenssen, H.; Moniri, M.R.; Hancock, R.E.W.; Pante, N. Bovine lactoferrin and lactoferricin interfere with intracellular trafficking of Herpes simplex virus-1. Biochimie 2009, 91, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, W.; Wakabayashi, H.; Takase, M.; Kawase, K.; Shimamura, S.; Tomita, M. Killing of Candida albicans by lactoferricin B, a potent antimicrobial peptide derived from the N-terminal region of bovine lactoferrin. Med. Microbiol. Immunol. (Berl) 1993, 182, 97–105. [Google Scholar] [CrossRef]
- Hug, D.H.; Hunter, J.K.; Dunkerson, D.D. Malnutrition, urocanic acid, and sun may interact to suppress immunity in sojourners to high altitude. Aviat. Space Environ. Med. 2001, 72, 136–145. [Google Scholar] [PubMed]
- Kolset, S.O.; Gallagher, J.T. Proteoglycans in haemopoietic cells. Biochim. Biophys. Acta 1990, 1032, 191–211. [Google Scholar] [PubMed]
- Iozzo, R.V.; Murdoch, A.D. Proteoglycans of the extracellular environment: clues from the gene and protein side offer novel perspectives in molecular diversity and function. Faseb J. 1996, 10, 598–614. [Google Scholar] [PubMed]
- Bernfield, M.; Kokenyesi, R.; Kato, M.; Hinkes, M.T.; Spring, J.; Gallo, R.L.; Lose, E.J. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu. Rev. Cell Biol. 1992, 8, 365–393. [Google Scholar] [PubMed]
- Parker, K.H.; Winlove, C.P.; Maroudas, A. The theoretical distributions and diffusivities of small ions in chondroitin sulphate and hyaluronate. Biophys. Chem. 1988, 32, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Iida, J.; Meijne, A.M.; Oegema Jr., T.R.; Yednock, T.A.; Kovach, N.L.; Furcht, L.T.; McCarthy, J.B. A role of chondroitin sulfate glycosaminoglycan binding site in alpha4beta1 integrin-mediated melanoma cell adhesion. J. Biol. Chem. 1998, 273, 5955–5962. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, I.; Kusche, M.; Unger, E.; Wlad, H.; Nylund, L.; Lindahl, U.; Kjellen, L. Biosynthesis of heparin. Purification of a 110-kDa mouse mastocytoma protein required for both glucosaminyl N-deacetylation and N-sulfation. J. Biol. Chem. 1991, 266, 8044–8049. [Google Scholar] [PubMed]
- Camejo, E.H.; Rosengren, B.; Camejo, G.; Sartipy, P.; Fager, G.; Bondjers, G. Interferon gamma binds to extracellular matrix chondroitin-sulfate proteoglycans, thus enhancing its cellular response. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1456–1465. [Google Scholar] [PubMed]
- Hoogewerf, A.J.; Kuschert, G.S.; Proudfoot, A.E.; Borlat, F.; Clark-Lewis, I.; Power, C.A.; Wells, T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. [Google Scholar] [CrossRef] [PubMed]
- Lookene, A.; Savonen, R.; Olivecrona, G. Interaction of lipoproteins with heparan sulfate proteoglycans and with lipoprotein lipase. Studies by surface plasmon resonance technique. Biochemistry 1997, 36, 5267–5275. [Google Scholar] [CrossRef] [PubMed]
- Olsson, U.; Camejo, G.; Hurt-Camejo, E.; Elfsber, K.; Wiklund, O.; Bondjers, G. Possible functional interactions of apolipoprotein B-100 segments that associate with cell proteoglycans and the ApoB/E receptor. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 149–155. [Google Scholar] [PubMed]
- Mettenleiter, T.C. Brief overview on cellular virus receptors. Virus Res. 2002, 82, 3–8. [Google Scholar] [CrossRef]
- Spillmann, D. Heparan sulfate: anchor for viral intruders? Biochimie 2001, 83, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Sandvik, K.; Andersen, J.H.; Hancock, R.E.W.; Gutteberg, T.J. Inhibition of HSV cell-to-cell spread by lactoferrin and lactoferricin. Antiviral Res. 2008, 79, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Song, B.H.; Lee, G.C.; Moon, M.S.; Cho, Y.H.; Lee, C.H. Human cytomegalovirus binding to heparan sulfate proteoglycans on the cell surface and/or entry stimulates the expression of human leukocyte antigen class I. J. Gen. Virol. 2001, 82, 2405–2413. [Google Scholar] [PubMed]
- Argyris, E.G.; Acheampong, E.; Nunnari, G.; Mukhtar, M.; Williams, K.J.; Pomerantz, R.J. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J. Virol. 2003, 77, 12140–12151. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Gibbs, B.F.; Toney, K.; Bennett, H.P. Purification of antimicrobial peptides from an extract of the skin of Xenopus laevis using heparin-affinity HPLC: characterization by ion-spray mass spectrometry. Anal. Biochem. 1994, 217, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Bjorck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Bjorck, L. Dermatan sulphate is released by proteinases of common pathogenic bacteria and inactivates antibacterial alpha-defensin. Mol. Microbiol. 2001, 39, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Romm, E.; Migliorini, M. Delineation of the glycosaminoglycan-binding site in the human inflammatory response protein lactoferrin. J. Biol. Chem. 1994, 269, 23661–23667. [Google Scholar] [PubMed]
- Shimazaki, K.; Tazume, T.; Uji, K.; Tanaka, M.; Kumura, H.; Mikawa, K.; Shimo-Oka, T. Properties of a heparin-binding peptide derived from bovine lactoferrin. J. Dairy Sci. 1998, 81, 2841–2849. [Google Scholar] [PubMed]
- Fromm, J.R.; Hileman, R.E.; Caldwell, E.E.; Weiler, J.M.; Linhardt, R.J. Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. Arch. Biochem. Biophys. 1995, 323, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hileman, R.E.; Fromm, J.R.; Weiler, J.M.; Linhardt, R.J. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 1998, 20, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Stenlund, P.; Lindberg, M.J.; Tibell, L.A. Structural requirements for high-affinity heparin binding: alanine scanning analysis of charged residues in the C-terminal domain of human extracellular superoxide dismutase. Biochemistry 2002, 41, 3168–3175. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Gutteberg, T.J.; Rekdal, O.; Lejon, T. Prediction of activity, synthesis and biological testing of anti-HSV active peptides. Chem. Biol. Drug Des. 2006, 68, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Hutton, R.D.; Ewert, D.L.; French, G.R. Differentiation of types 1 and 2 herpes simplex virus by plaque inhibition with sulfated polyanions. Proc. Soc. Exp. Biol. Med. 1973, 142, 27–29. [Google Scholar] [PubMed]
- Langeland, N.; Moore, L.J.; Holmsen, H.; Haarr, L. Interaction of polylysine with the cellular receptor for herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim Biophys Acta 1999, 1462, 29–54. [Google Scholar] [CrossRef] [PubMed]
- Lorin, C.; Saidi, H.; Belaid, A.; Zairi, A.; Baleux, F.; Hocini, H.; Belec, L.; Hani, K.; Tangy, F. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology 2005, 334, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Niwa, M.; Tokunaga, F.; Miyata, T.; Iwanaga, S. Direct virus inactivation of tachyplesin I and its isopeptides from horseshoe crab hemocytes. Chemotherapy 1991, 37, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.; Li, Q.X.; Perez-Romero, P.; Delassus, G.; Lopez, S.R.; Sutter, S.; McLaren, N.; Fuller, A.O. A new class of receptor for herpes simplex virus has heptad repeat motifs that are common to membrane fusion proteins. J. Virol. 2005, 79, 7419–7430. [Google Scholar] [CrossRef] [PubMed]
- Perez-Romero, P.; Fuller, A.O. The C terminus of the B5 receptor for herpes simplex virus contains a functional region important for infection. J. Virol. 2005, 79, 7431–7437. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Scott, M.G.; Hancock, R.E.W. Immunomodulatory activities of small host defense peptides. Antimicrob. Agents Chemother. 2005, 49, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Javadpour, M.M.; Barkley, M.D. Self-assembly of designed antimicrobial peptides in solution and micelles. Biochemistry 1997, 36, 9540–9549. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.L.; Vargas Jr., J.; DelPortillo, A.; Klotman, M.E. Dual role of alpha-defensin-1 in anti-HIV-1 innate immunity. J. Clin. Invest. 2005, 115, 765–773. [Google Scholar] [PubMed]
- Futaki, S.; Nakase, I.; Suzuki, T.; Youjun, Z.; Sugiura, Y. Translocation of branched-chain arginine peptides through cell membranes: flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry 2002, 41, 7925–7930. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S. Arginine-rich peptides: potential for intracellular delivery of macromolecules and the mystery of the translocation mechanisms. Int. J. Pharm. 2002, 245, 1–7. [Google Scholar] [CrossRef]
- Suzuki, T.; Futaki, S.; Niwa, M.; Tanaka, S.; Ueda, K.; Sugiura, Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J. Biol. Chem. 2002, 277, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Penco, S.; Scarfi, S.; Giovine, M.; Damonte, G.; Millo, E.; Villaggio, B.; Passalacqua, M.; Pozzolini, M.; Garre, C.; Benatti, U. Identification of an import signal for, and the nuclear localization of, human lactoferrin. Biotechnol. Appl. Biochem. 2001, 34, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef] [PubMed]
- Kanyshkova, T.G.; Semenov, D.V.; Buneva, V.N.; Nevinsky, G.A. Human milk lactoferrin binds two DNA molecules with different affinities. FEBS Lett. 1999, 451, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.M.; Park, Y.; Lim, S.S.; Yang, S.T.; Woo, E.R.; Park, I.S.; Lee, J.S.; Kim, J.I.; Hahm, K.S.; Kim, Y.; Shin, S.Y. Cell selectivity and mechanism of action of antimicrobial model peptides containing peptoid residues. Biochemistry 2005, 44, 12094–12106. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, S.; Wittrup, A.; Cheng, F.; Jonsson, M.; Eklund, E.; Busch, S.; Belting, M. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J. Biol. Chem. 2004, 279, 17951–17956. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R.E.W. Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E.W. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [PubMed]
- Luebke, R.W.; Parks, C.; Luster, M.I. Suppression of immune function and susceptibility to infections in humans: association of immune function with clinical disease. J. Immunotoxicol. 2004, 1, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Someya, A.; Hirata, M.; Ogawa, H.; Nagaoka, I. Evaluation of the effects of peptide antibiotics human beta-defensins-1/-2 and LL-37 on histamine release and prostaglandin D(2) production from mast cells. Eur. J. Immunol. 2001, 31, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Lande, R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr. Opin. Immunol. 2008, 20, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Fellermann, K.; Herrlinger, K.R.; Baxmann, S.; Schmidt, K.; Schwind, B.; Duchrow, M.; Wohlschlager, C.; Feller, A.C.; Stange, E.F. Human beta-defensin 2 but not beta-defensin 1 is expressed preferentially in colonic mucosa of inflammatory bowel disease. Eur J. Gastroenterol. Hepatol. 2002, 14, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Harder, J.; Weichenthal, M.; Mueller, O.; Herrlinger, K.R.; Fellermann, K.; Schroeder, J.M.; Stange, E.F. Inducible and constitutive beta-defensins are differentially expressed in Crohn's disease and ulcerative colitis. Inflamm. Bowel Dis. 2003, 9, 215–223. [Google Scholar] [CrossRef]
- Schauber, J.; Rieger, D.; Weiler, F.; Wehkamp, J.; Eck, M.; Fellermann, K.; Scheppach, W.; Gallo, R.L.; Stange, E.F. Heterogeneous expression of human cathelicidin hCAP18/LL-37 in inflammatory bowel diseases. Eur J. Gastroenterol. Hepatol. 2006, 18, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Schmid, M.; Stange, E.F. Defensins and other antimicrobial peptides in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2007, 23, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Biron, E.; Chatterjee, J.; Ovadia, O.; Langenegger, D.; Brueggen, J.; Hoyer, D.; Schmid, H.A.; Jelinek, R.; Gilon, C.; Hoffman, A.; Kessler, H. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew. Chem. Int. Ed. Engl. 2008, 47, 2595–2599. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-methylation of peptides: a new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L. Pegasys (Hoffmann-La Roche). Curr. Opin. Investig. Drugs 2001, 2, 1530–1538. [Google Scholar] [PubMed]
- Davidson, D.J.; Currie, A.J.; Reid, G.S.; Bowdish, D.M.; MacDonald, K.L.; Ma, R.C.; Hancock, R.E.W.; Speert, D.P. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J. Immunol. 2004, 172, 1146–1156. [Google Scholar] [PubMed]
- Schijns, V.E. Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 2000, 12, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Johnson, T.R.; Peebles, R.S. Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology 2000, 48, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Waris, M.E.; Tsou, C.; Erdman, D.D.; Zaki, S.R.; Anderson, L.J. Respiratory synctial virus infection in BALB/c mice previously immunized with formalin-inactivated virus induces enhanced pulmonary inflammatory response with a predominant Th2-like cytokine pattern. J. Virol. 1996, 70, 2852–2860. [Google Scholar] [PubMed]
- Kovacs-Nolan, J.; Mapletoft, J.W.; Lawman, Z.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Formulation of bovine respiratory syncytial virus fusion protein with CpG oligodeoxynucleotide, cationic host defence peptide and polyphosphazene enhances humoral and cellular responses and induces a protective type 1 immune response in mice. J. Gen. Virol. 2009, 90, 1892–1905. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Nolan, J.; Mapletoft, J.W.; Latimer, L.; Babiuk, L.A.; Hurk, S.D. CpG oligonucleotide, host defense peptide and polyphosphazene act synergistically, inducing long-lasting, balanced immune responses in cattle. Vaccine 2009, 27, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- Kovacs-Nolan, J.; Latimer, L.; Landi, A.; Jenssen, H.; Hancock, R.E.W.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. The novel adjuvant combination of CpG ODN, indolicidin and polyphosphazene induces potent antibody- and cell-mediated immune responses in mice. Vaccine 2009, 27, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Jenssen, H.; Elliott, M.; Townsend, R.; Nijnik, A.; Lee, S.F.; Gerdts, V.; Babiuk, L.A.; Halperin, S.A.; Hancock, R.E.W. A novel vaccine adjuvant comprised of a synthetic innate defence regulator peptide and CpG oligonucleotide links innate and adaptive immunity. Vaccine 2009, 27, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Fritz, J.H.; Brunner, S.; Birnstiel, M.L.; Buschle, M.; Gabain, A.; Mattner, F.; Zauner, W. The artificial antimicrobial peptide KLKLLLLLKLK induces predominantly a TH2-type immune response to co-injected antigens. Vaccine 2004, 22, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Lingnau, K.; Egyed, A.; Schellack, C.; Mattner, F.; Buschle, M.; Schmidt, W. Poly-L-arginine synergizes with oligodeoxynucleotides containing CpG-motifs (CpG-ODN) for enhanced and prolonged immune responses and prevents the CpG-ODN-induced systemic release of pro-inflammatory cytokines. Vaccine 2002, 20, 3498–3508. [Google Scholar] [CrossRef] [PubMed]
- Aichinger, M.C.; Ortbauer, M.; Reipert, S.; Zauner, W.; Bogner, P.; Froschauer, E.; Nowikovsky, K.; Lingnau, K.; von Gabain, A.; Schweyen, R.; Henics, T. Unique membrane-interacting properties of the immunostimulatory cationic peptide KLKL(5)KLK (KLK). Cell Biol. Int. 2008, 32, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Schellack, C.; Prinz, K.; Egyed, A.; Fritz, J.H.; Wittmann, B.; Ginzler, M.; Swatosch, G.; Zauner, W.; Kast, C.; Akira, S.; von Gabain, A.; Buschle, M.; Lingnau, K. IC31, a novel adjuvant signaling via TLR9, induces potent cellular and humoral immune responses. Vaccine 2006, 24, 5461–5472. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Blomberg, L.; Flegel, M.; Lepsa, L.; Nilsson, B.; Verlander, M. Large-scale synthesis of peptides. Biopolymers 2000, 55, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.E.; Bray, B.L.; Mader, C.J.; Friedrich, P.E.; Anderson, M.W.; Taylor, T.S.; Boshernitzan, N.; Niemi, T.E.; Fulcher, B.C.; Whight, S.R.; White, J.M.; Greene, R.J.; Stoltenberg, L.E.; Lichty, M. Development of HIV fusion inhibitors. J. Pept. Sci. 2005, 11, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, C.T.; Thomsen, L.E.; Ingmer, H.; Mygind, P.H.; Kristensen, H.H.; Gram, L. Antimicrobial peptides effectively kill a broad spectrum of Listeria monocytogenes and Staphylococcus aureus strains independently of origin, sub-type, or virulence factor expression. BMC Microbiol. 2008, 8, 205. [Google Scholar] [CrossRef]
- Goldstein, A.L.; Low, T.L.; McAdoo, M.; McClure, J.; Thurman, G.B.; Rossio, J.; Lai, C.Y.; Chang, D.; Wang, S.S.; Harvey, C.; Ramel, A.H.; Meienhofer, J. Thymosin alpha1: isolation and sequence analysis of an immunologically active thymic polypeptide. Proc. Natl. Acad. Sci. U S A 1977, 74, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rasi, G.; Pierimarchi, P.; Sinibaldi Vallebona, P.; Colella, F.; Garaci, E. Combination therapy in the treatment of chronic viral hepatitis and prevention of hepatocellular carcinoma. Int Immunopharmacol 2003, 3, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Khavinson, V.K.; Morozov, V.G. Immunomodulatory synthetic dipeptide L-Glu-L-Trp slows down aging and inhibits spontaneous carcinogenesis in rats. Biogerontology 2000, 1, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Tulpule, A.; Scadden, D.T.; Espina, B.M.; Cabriales, S.; Howard, W.; Shea, K.; Gill, P.S. Results of a randomized study of IM862 nasal solution in the treatment of AIDS-related Kaposi's sarcoma. J. Clin. Oncol. 2000, 18, 716–723. [Google Scholar] [PubMed]
- Bergeon, J.A.; Toth, I. Enhancement of oral drug absorption-effect of lipid conjugation on the enzymatic stability and intestinal permeability of l-Glu-l-Trp-NH(2). Bioorg. Med. Chem. 2007, 15, 7048–7057. [Google Scholar] [CrossRef] [PubMed]
- Rose 2nd, W.A.; Tuthill, C.; Pyles, R.B. An immunomodulating dipeptide, SCV-07, is a potential therapeutic for recurrent genital herpes simplex virus type 2 (HSV-2). Int. J. Antimicrob. Agents 2008, 32, 262–266. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Jenssen, H. Therapeutic Approaches Using Host Defence Peptides to Tackle Herpes Virus Infections. Viruses 2009, 1, 939-964. https://doi.org/10.3390/v1030939
Jenssen H. Therapeutic Approaches Using Host Defence Peptides to Tackle Herpes Virus Infections. Viruses. 2009; 1(3):939-964. https://doi.org/10.3390/v1030939
Chicago/Turabian StyleJenssen, Håvard. 2009. "Therapeutic Approaches Using Host Defence Peptides to Tackle Herpes Virus Infections" Viruses 1, no. 3: 939-964. https://doi.org/10.3390/v1030939
APA StyleJenssen, H. (2009). Therapeutic Approaches Using Host Defence Peptides to Tackle Herpes Virus Infections. Viruses, 1(3), 939-964. https://doi.org/10.3390/v1030939