Activation and Evasion of Innate Antiviral Immunity by Herpes Simplex Virus

Abstract
1. Introduction
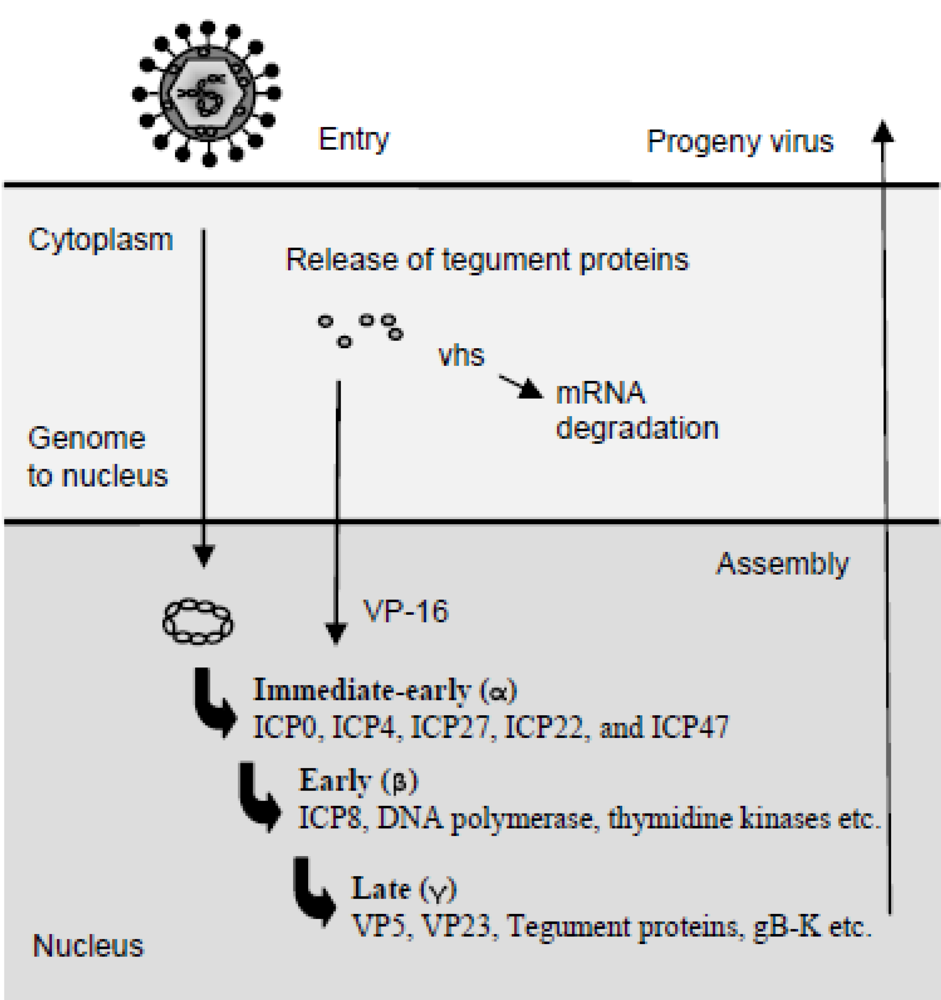
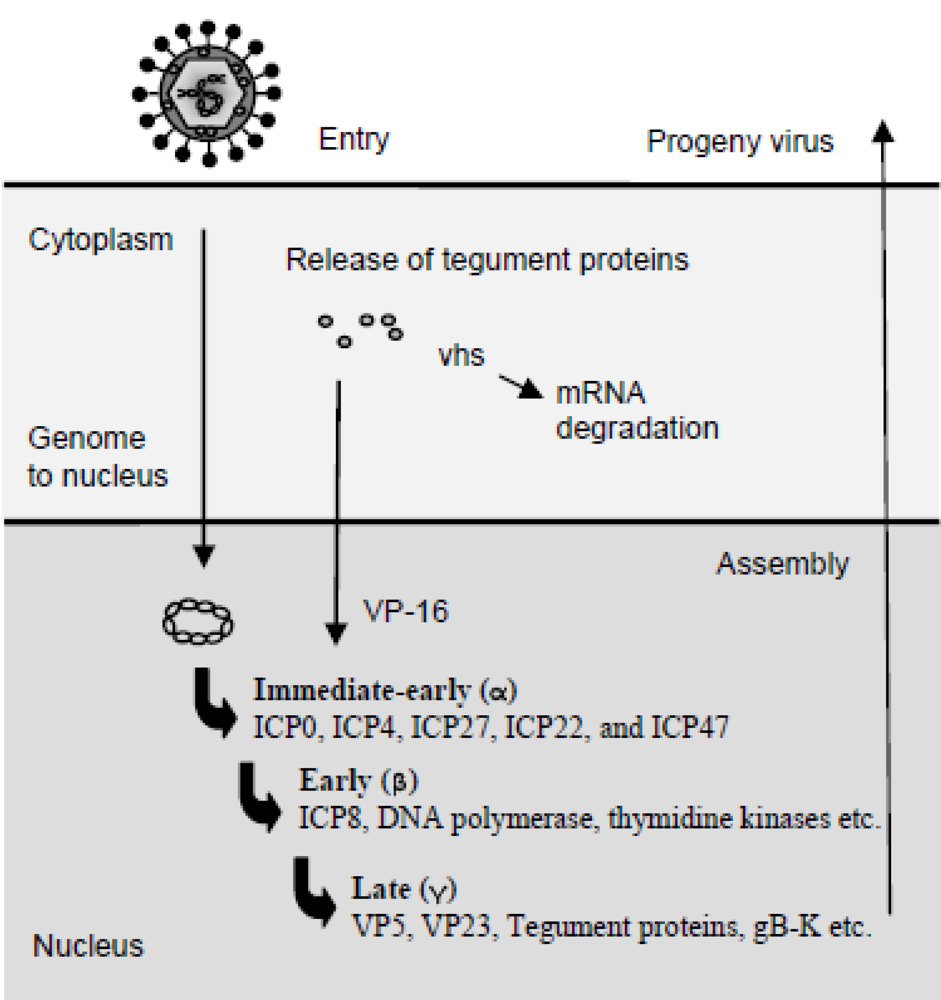
2. Herpes Simplex Virus and Replication of the Virus Genome
3. Antiviral IFN Response during Early HSV Infection
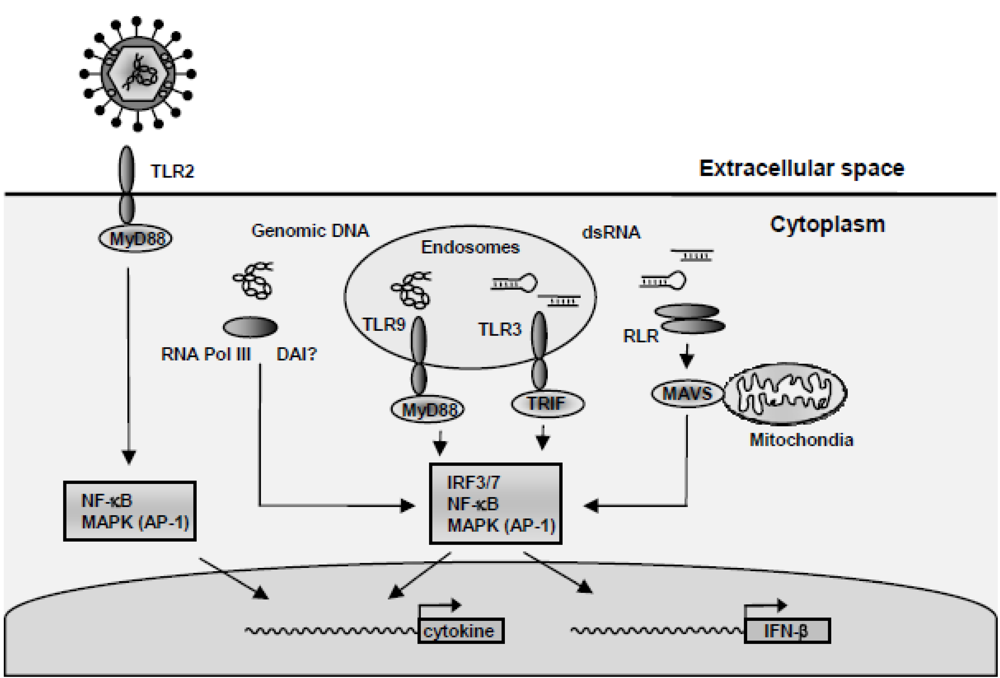
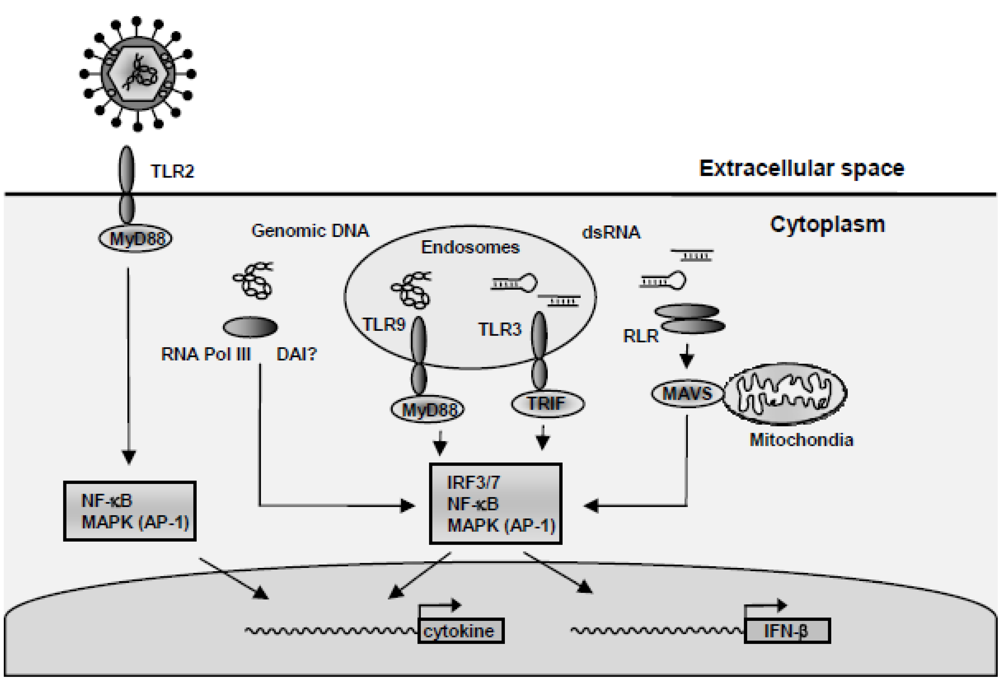
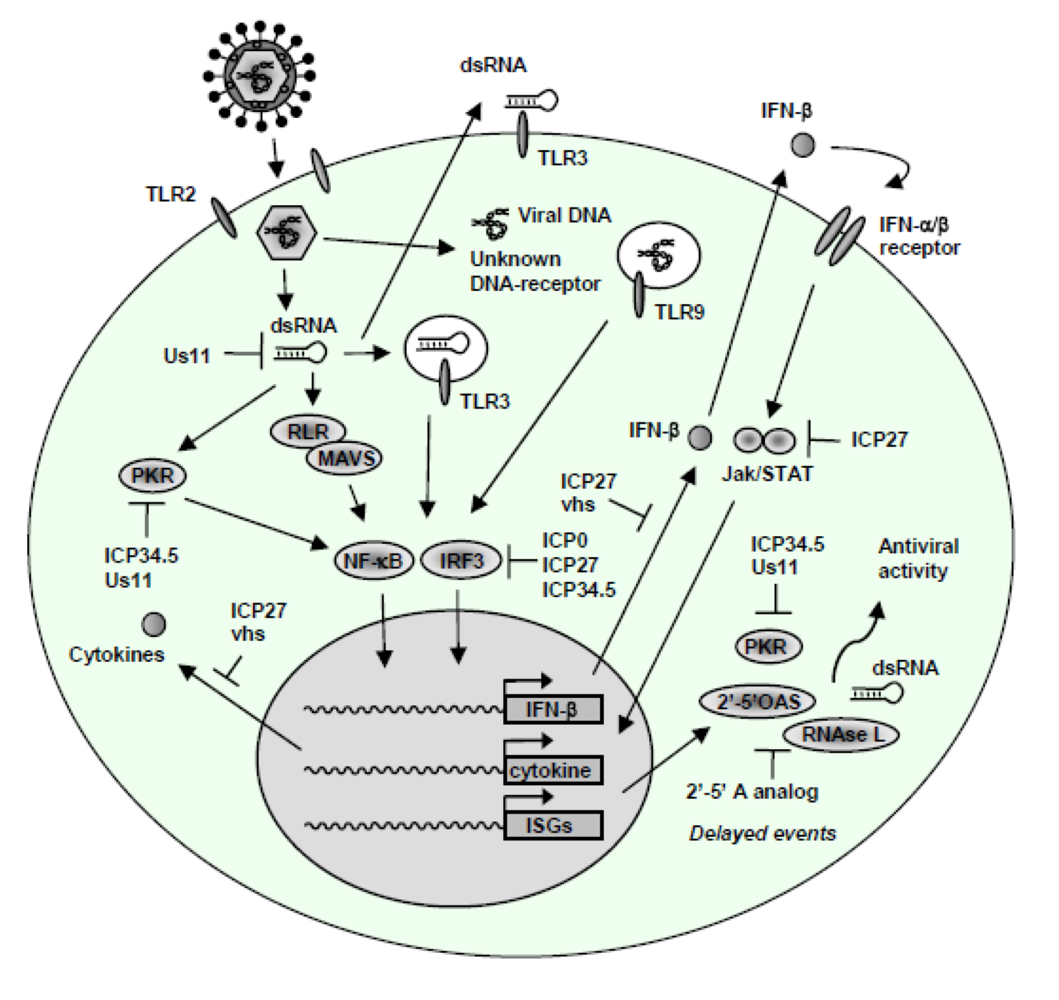
4. HSV-Activated Signaling Pathways
5. Evasion of the Innate Immune System by HSV
5.1 Evasion of IFN signalling and IFN effector functions


{kind=link}
{kind=link}
{kind=link}
| Viral protein | Effect | Mechanism | References |
|---|---|---|---|
| Inhibition of IFN and cytokine signalling and IFN function | |||
| ICP0 | Enhanced resistance to IFN | Modification of IRF3 and IRF7 activation | [70-74] |
| ICP27 | Decreased IFN and cytokine expression | Reduces IRF3 and NF-κB activation | [27] |
| Us3 | Decreased ISG expression (Mx) and reduced IRF3 activation | n.d. | [79] |
| vhs | Inhibition of IFN-α/β production | Potentially because of reduced IRF7 activation | [80-82] |
| vhs | Inhibition of JAK/STAT signalling | Induction of SOCS3, repression of STAT1 activation | [82,83] |
| ICP27 | Inhibition of IFN signalling | Decreased STAT1 activation and translocation to the nucleus | [78] |
| vhs | Suppression of proinflammatory cytokines, IFNs, and chemokines | n.d. | [84] |
| ICP34.5 | Suppression of antiviral genes | Inhibition of IRF3 activation via interaction with TBK1 | [85] |
| ICP34.5 | Inhibition of PKR and PERK activity | Reverses the PKR and PERK-induced phosphorylation of eIF2α | [61,62,86] |
| Us11 | Inhibition of dsRNA-dependent and PACT-mediated activation of PKR | Binds to dsRNABinds to PKR | [63][64] |
| Us11 | Inhibition of 2’-5’ OAS | Binding to dsRNA (dsRNA binding domain of Us11 essential) | [67] |
| 2’-5’A analog | Inhibition of the 2’-5’ OAS/ RNAse L system | 2’-5’ A analogue | [69] |
| ICP0 | Inhibition of RNAseL-independent rRNA degradation | n.d. | [68] |
| Inhibition of host gene expression | |||
| ICP0 | Inhibition of TLR-induced JNK and NF-κB activation | Recruitment of USP7 binding to TRAF6 and IKKγ | [87] |
| ICP27 | Inhibition of splicing | Interacts with spliceosome components | [88-90] |
| ICP27 | Reduction of mRNA stability | n.d. | [91] |
| VP16 and vhs | RNA degradation | n.d. | [92] |
| ICP0 | Cell cycle arrest and disturbed cellular gene expression | Upregulation of p53-responsive genes. | [93] |
| Unknown | Inhibition of NFAT activation | n.d. | [94] |
| Inhibition of apoptosis | |||
| ICP4 | Inhibition of apoptosis | n.d. | [95] |
| gJ | Inhibition of apoptosis | Inhibition of caspase activation | [96] |
| ICP27 | Inhibition of apoptosis | n.d. | [97] |
| ICP34.5 | Inhibition of apoptosis | Inhibition of PKR activity | [62] |
| Inhibition of CTL-induced cell death (apoptosis) | Downregulation of cell surface Fas ligand | [98,99] | |
| Inhibition of autophagy and anti-microbial proteins | |||
| ICP0 and ICP0 | Inhibition of SLPI | n.d. | [100] |
| ICP34.5 | Inhibition of autophagy | Targeting of Beclin-1 | [66] |
| Inhibition of complement, antigen presentation and APC function | |||
| gC | Inhibition of complement | Binds to complement factor C3 | [101,102] |
| gE/gI complex | Blocking of Fc-mediated activities, including complement activation and ADCC | Binds to Fc domain of IgG | [103] |
| vhs | Inhibition of DC maturation and reduced cytokine production | n.d. | [104,105] |
| ICP47 | Inhibition of antigen presentation by MHC I | Interferes with TAP1/TAP2 | [106-108] |
| vhs | Inhibition of antigen presentation by MHC I and MHC II | Interferes with MHC I transport. Reduces levels of MHC II | [109-111] |
| gB | Inhibition of MHC II-mediated antigen presentation | Inhibited expression of invariant chain and interacts with HLA-DR and HLA-DM | [112] |
5.2 Inhibition of cytokine and IFN gene transcription and translation
5.3 Inhibition of autophagy and intrinsic protection
6. Concluding remarks
References and Notes
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex virus. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2007; Volume 2, pp. 2501–2601. [Google Scholar]
- Pinto, A.J.; Morahan, P.S.; Brinton, M.; Stewart, D.; Gavin, E. Comparative therapeutic efficacy of recombinant interferons-alpha, -beta, and -gamma against alphatogavirus, bunyavirus, flavivirus, and herpesvirus infections. J. Interferon Res. 1990, 10, 293–298. [Google Scholar] [PubMed]
- Leib, D.A.; Harrison, T.E.; Laslo, K.M.; Machalek, M.A.; Moorman, N.J.; Virgin, H.W. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 1999, 189, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Jouanguy, E.; Al Hajjar, S.; Fieschi, C.; Al Mohsen, I.Z.; Al Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; Al Ghonaium, A.; Tufenkeji, H.; Frayha, H.; Al Gazlan, S.; Al Rayes, H.; Schreiber, R.D.; Gresser, I.; Casanova, J.L. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; Staeheli, P.; Hartmann, R.; Jensen, U.B.; Dagnaes-Hansen, F.; Thomsen, A.R.; Chen, Z.; Haugen, H.; Klucher, K.; Paludan, S.R. An important role for type III Interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar] [PubMed]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; Ohba, Y.; Taniguchi, T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.H.; Macmillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Palmquist, J.M.; Lokensgard, J.R. Cutting Edge: TLR2-Mediated Proinflammatory Cytokine and Chemokine Production by Microglial Cells in Response to Herpes Simplex Virus. J. Immunol. 2005, 175, 4189–4193. [Google Scholar] [PubMed]
- Reske, A.; Pollara, G.; Krummenacher, C.; Katz, D.R.; Chain, B.M. Glycoprotein-dependent and TLR2-independent innate immune recognition of herpes simplex virus-1 by dendritic cells. J. Immunol. 2008, 180, 7525–7536. [Google Scholar] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Hochrein, H.; Schlatter, B.; O'Keeffe, M.; Wagner, C.; Schmitz, F.; Schiemann, M.; Bauer, S.; Suter, M.; Wagner, H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2004, 101, 11416–11421. [Google Scholar] [CrossRef]
- Malmgaard, L.; Melchjorsen, J.; Bowie, A.G.; Mogensen, S.C.; Paludan, S.R. Viral activation of macrophages through TLR-dependent and -independent pathways. J. Immunol. 2004, 173, 6890–6898. [Google Scholar] [PubMed]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I IFN production during herpes simplex virus infection is controlled by cell-type specific viral recognition through TLR9, the MAVS pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Picard, C.; Chapgier, A.; Plancoulaine, S.; Titeux, M.; Cognet, C.; von Bernuth, H.; Ku, C.L.; Casrouge, A.; Zhang, X.X.; Barreiro, L.; Leonard, J.; Hamilton, C.; Lebon, P.; Heron, B.; Vallee, L.; Quintana-Murci, L.; Hovnanian, A.; Rozenberg, F.; Vivier, E.; Geissmann, F.; Tardieu, M.; Abel, L.; Casanova, J.L. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Dolan, A.; Jamieson, F.E.; Cunningham, C.; Barnett, B.C.; McGeoch, D.J. The genome sequence of herpes simplex virus type 2. J. Virol. 1998, 72, 2010–2021. [Google Scholar] [PubMed]
- Fruh, K.; Gruhler, A.; Krishna, R.M.; Schoenhals, G.J. A comparison of viral immune escape strategies targeting the MHC class I assembly pathway. Immunol. Rev. 1999, 168, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Homa, F.L.; Brown, J.C. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 1997, 7, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Ellermann-Eriksen, S. Autocrine secretion of interferon-alpha/beta and tumour necrosis factor- alpha synergistically activates mouse macrophages after infection with herpes simplex virus type 2. J. Gen. Virol. 1993, 74, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Mittnacht, S.; Straub, P.; Kirchner, H.; Jacobsen, H. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology 1988, 164, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Oberman, F.; Panet, A. Inhibition of transcription of herpes simplex virus immediate early genes in interferon-treated human cells. J. Gen. Virol. 1988, 69, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; Halford, W.P. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1 . J. Virol. 2002, 76 , 11541–11550. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, S.; Arnold, S.; Schwerdel, C.; Franchini, M.; Alber, G.; Di Santo, J.P.; Ackermann, M.; Suter, M. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J. Virol. 2004, 78, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Gresser, I.; Tovey, M.G.; Maury, C.; Bandu, M.T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J. Exp. Med. 1976, 144, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Siren, J.; Julkunen, I.; Paludan, S.R.; Matikainen, S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen. Virol. 2006, 87, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Khabar, K.S.; Dhalla, M.; Siddiqui, Y.; Zhou, A.; Al Ahdal, M.N.; Der, S.D.; Silverman, R.H.; Williams, B.R. Effect of deficiency of the double-stranded RNA-dependent protein kinase, PKR, on antiviral resistance in the presence or absence of ribonuclease L: HSV-1 replication is particularly sensitive to deficiency of the major IFN-mediated enzymes. J. Interferon Cytokine Res. 2000, 20, 653–659. [Google Scholar] [PubMed]
- Melchjorsen, J.; Pedersen, F.S.; Mogensen, S.C.; Paludan, S.R. Herpes simplex virus selectively induces expression of the CC Chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J. Virol. 2002, 76, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Silverman, R.H.; Zhou, A.; Goto, T.; Kwon, B.S.; Kaufman, H.E.; Hill, J.M. Increased severity of HSV-1 keratitis and mortality in mice lacking the 2-5A-dependent RNase L gene. Invest Ophthalmol. Vis. Sci. 2001, 42, 120–126. [Google Scholar] [PubMed]
- Carr, D.J.; Al khatib, K.; James, C.M.; Silverman, R. Interferon-beta suppresses herpes simplex virus type 1 replication in trigeminal ganglion cells through an RNase L-dependent pathway. J. Neuroimmunol. 2003, 141, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, M.; Milligan, J.R.; Kaji, A. Effect of 2',5'-oligoadenylate on herpes simplex virus-infected cells and preventive action of 2',5'-oligoadenylate on the lethal effect of HSV-2. J. Interferon Res. 1989, 9, 691–707. [Google Scholar] [CrossRef] [PubMed]
- Neville, L.F.; Mathiak, G.; Bagasra, O. The immunobiology of interferon-gamma inducible protein 10 kD (IP-10): a novel, pleiotropic member of the C-X-C chemokine superfamily. Cytokine Growth Factor Rev. 1997, 8, 207–219. [Google Scholar] [CrossRef]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [PubMed]
- Miettinen, M.; Sareneva, T.; Julkunen, I.; Matikainen, S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2001, 2, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Siren, J.; Pirhonen, J.; Julkunen, I.; Matikainen, S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J. Immunol. 2005, 174, 1932–1937. [Google Scholar] [PubMed]
- Foy, E.; Li, K.; Sumpter, R.; Loo, Y.M.; Johnson, C.L.; Wang, C.; Fish, P.M.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling . Proc. Natl. Acad. Sci. USA 2005, 102 , 2986–2991. [Google Scholar] [CrossRef]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN-{lambda}), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; Ostrander, C.; Dong, D.; Shin, J.; Presnell, S.; Fox, B.; Haldeman, B.; Cooper, E.; Taft, D.; Gilbert, T.; Grant, F.J.; Tackett, M.; Krivan, W.; McKnight, G.; Clegg, C.; Foster, D.; Klucher, K.M. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.M.; Severa, M.; Giacomini, E.; Monneron, D.; Remoli, M.E.; Julkunen, I.; Cella, M.; Lande, R.; Uze, G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 2004, 34, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Karupiah, G.; Xie, Q.W.; Buller, R.M.; Nathan, C.; Duarte, C.; MacMicking, J.D. Inhibition of viral replication by interferon-gamma-induced nitric oxide synthase. Science 1993, 261, 1445–1448. [Google Scholar] [PubMed]
- Chesler, D.A.; Reiss, C.S. The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002, 13, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, S.; Franchini, M.; Alber, G.; Ackermann, M.; Suter, M. Interleukin-12- and gamma interferon-dependent innate immunity are essential and sufficient for long-term survival of passively immunized mice infected with herpes simplex virus type 1. J. Virol. 2001, 75, 9596–9600. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.C. Viruses and interferons. Annu. Rev. Microbiol. 2001, 55, 255–281. [Google Scholar] [CrossRef] [PubMed]
- Malmgaard, L.; Paludan, S.R. Interferon (IFN)-alpha/beta, interleukin (IL)-12 and IL-18 coordinately induce production of IFN-gamma during infection with herpes simplex virus type 2. J. Gen. Virol. 2003, 84, 2497–2500. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Mallo, M.; Eichmann, K.; Modolell, M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J. Exp. Med. 1998, 187, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Schindler, H.; Lutz, M.B.; Rollinghoff, M.; Bogdan, C. The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J. Immunol. 2001, 166, 3075–3082. [Google Scholar] [PubMed]
- Stober, D.; Schirmbeck, R.; Reimann, J. IL-12/IL-18-dependent IFN-gamma release by murine dendritic cells. J. Immunol. 2001, 167, 957–965. [Google Scholar] [PubMed]
- Kodukula, P.; Liu, T.; Rooijen, N.V.; Jager, M.J.; Hendricks, R.L. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J. Immunol. 1999, 162, 2895–2905. [Google Scholar] [PubMed]
- Paludan, S.R.; Mogensen, S.C. Virus-cell interactions regulating induction of tumor necrosis factor alpha production in macrophages infected with herpes simplex virus. J. Virol. 2001, 75, 10170–10178. [Google Scholar] [CrossRef] [PubMed]
- Malmgaard, L.; Paludan, S.R.; Mogensen, S.C.; Ellermann-Eriksen, S. Herpes simplex virus type 2 induces secretion of IL-12 by macrophages through a mechanism involving NF-kappaB. J. Gen. Virol. 2000, 81, 3011–3020. [Google Scholar] [PubMed]
- Imaizumi, T.; Hatakeyama, M.; Yamashita, K.; Yoshida, H.; Ishikawa, A.; Taima, K.; Satoh, K.; Mori, F.; Wakabayashi, K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothelium 2004, 11, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Jensen, S.B.; Nielsen, C.; Quartin, E.; Kato, H.; Chen, Z.J.; Silverman, R.H.; Akira, S.; Paludan, S.R. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J. Gen. Virol. 2009, 90, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hanson, J.; McLean, T.I.; Olgiate, J.; Hilton, M.; Miller, W.E.; Bachenheimer, S.L. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 1998, 247, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; O'Shea, J.J. Jaks and STATs: biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef] [PubMed]
- Pasieka, T.J.; Lu, B.; Leib, D.A. Enhanced pathogenesis of an attenuated herpes simplex virus for mice lacking Stat1. J. Virol. 2008, 82, 6052–6055. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1 . Proc. Natl. Acad. Sci. USA 1995, 92 , 10516–10520. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase . Proc. Natl. Acad. Sci. USA 1997, 94 , 843–848. [Google Scholar] [CrossRef]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [PubMed]
- Mulvey, M.; Camarena, V.; Mohr, I. Full resistance of herpes simplex virus type 1-infected primary human cells to alpha interferon requires both the Us11 and gamma(1)34.5 gene products . J. Virol. 2004, 78, 10193–10196. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host. Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Sanchez, R.; Mohr, I. Inhibition of cellular 2'-5' oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 2007, 81, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Sobol, P.T.; Mossman, K.L. ICP0 Prevents RNase L-Independent rRNA Cleavage in Herpes Simplex Virus Type 1-Infected Cells. J. Virol. 2006, 80, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Cayley, P.J.; Davies, J.A.; McCullagh, K.G.; Kerr, I.M. Activation of the ppp(A2'p)nA system in interferon-treated, herpes simplex virus-infected cells and evidence for novel inhibitors of the ppp(A2'p)nA-dependent RNase. Eur.J.Biochem. 1984, 143, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000, 74, 2052–2056. [Google Scholar] [CrossRef] [PubMed]
- Harle, P.; Sainz, B.; Carr, D.J.; Halford, W.P. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta . Virology 2002, 293, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3 and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Melroe, G.T.; DeLuca, N.A.; Knipe, D.M. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 2004, 78, 8411–8420. [Google Scholar] [CrossRef] [PubMed]
- Eidson, K.M.; Hobbs, W.E.; Manning, B.J.; Carlson, P.; DeLuca, N.A. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral Infection. J. Virol. 2002, 76, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Halford, W.P.; Weisend, C.; Grace, J.; Soboleski, M.; Carr, D.J.; Balliet, J.W.; Imai, Y.; Margolis, T.P.; Gebhardt, B.M. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol. J. 2006, 3, 44. [Google Scholar] [CrossRef]
- Everett, R.D.; Orr, A. Herpes simplex virus type 1 regulatory protein ICP0 aids infection in cells with a preinduced interferon response but does not impede interferon-induced gene induction. J. Virol. 2009, 83, 4978–4983. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Young, D.F.; Randall, R.E.; Orr, A. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 2008, 82, 8871–8881. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.A.; Duerst, R.J.; Smith, T.J.; Morrison, L.A. Herpes simplex virus type 2 virion host shutoff protein regulates alpha/beta interferon but not adaptive immune responses during primary infection in vivo. J. Virol. 2003, 77, 9337–9345. [Google Scholar] [CrossRef] [PubMed]
- Duerst, R.J.; Morrison, L.A. Herpes simplex virus 2 virion host shutoff protein interferes with type I interferon production and responsiveness. Virology 2004, 322, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yokosawa, N.; Okabayashi, T.; Suzutani, T.; Miura, S.; Jimbow, K.; Fujii, N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 2004, 78, 6282–6286. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Yokosawa, N.; Kubota, T.; Suzutani, T.; Yoshida, I.; Miura, S.; Jimbow, K.; Fujii, N. Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and janus kinases during an early infection stage. Virology 2001, 286, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Suzutani, T.; Nagamine, M.; Shibaki, T.; Ogasawara, M.; Yoshida, I.; Daikoku, T.; Nishiyama, Y.; Azuma, M. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J. Gen. Virol. 2000, 81, 1763–1771. [Google Scholar] [PubMed]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1 . J. Biol. Chem. 2009, 284 , 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein . J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Hardy, W.R.; Sandri-Goldin, R.M. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 1994, 68, 7790–7799. [Google Scholar] [PubMed]
- Sciabica, K.S.; Dai, Q.J.; Sandri-Goldin, R.M. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J. 2003, 22, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Wadd, S.E.; Lamond, A.I.; Silverstein, S.J.; Clements, J.B. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J. Virol. 2001, 75, 4376–4385. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Melchjorsen, J.; Malmgaard, L.; Casola, A.; Paludan, S.R. Suppression of proinflammatory cytokine expression by herpes simplex virus type 1. J. Virol. 2004, 78, 5883–5890. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.S.; Leib, D.A. Role of the VP16-binding domain of vhs in viral growth, host shutoff activity, and pathogenesis. J. Virol. 2004, 78, 13562–13572. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, W.E.; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar] [PubMed]
- Scott, E.S.; Malcomber, S.; O'Hare, P. Nuclear translocation and activation of the transcription factor NFAT is blocked by herpes simplex virus infection. J. Virol. 2001, 75, 9955–9965. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, R.; Roizman, B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc. Natl. Acad. Sci. USA 1996, 93, 9583–9587. [Google Scholar] [CrossRef]
- Jerome, K.R.; Chen, Z.; Lang, R.; Torres, M.R.; Hofmeister, J.; Smith, S.; Fox, R.; Froelich, C.J.; Corey, L. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or Fas. J. Immunol. 2001, 167, 3928–3935. [Google Scholar] [PubMed]
- Aubert, M.; Blaho, J.A. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 1999, 73, 2803–2813. [Google Scholar] [PubMed]
- Sieg, S.; Yildirim, Z.; Smith, D.; Kayagaki, N.; Yagita, H.; Huang, Y.; Kaplan, D. Herpes simplex virus type 2 inhibition of Fas ligand expression. J. Virol. 1996, 70, 8747–8751. [Google Scholar] [PubMed]
- Sieg, S.; Huang, Y.; Kaplan, D. Viral regulation of CD95 expression and apoptosis in T lymphocytes. J. Immunol. 1997, 159, 1192–1199. [Google Scholar] [PubMed]
- Fakioglu, E.; Wilson, S.S.; Mesquita, P.M.; Hazrati, E.; Cheshenko, N.; Blaho, J.A.; Herold, B.C. Herpes simplex virus downregulates secretory leukocyte protease inhibitor: a novel immune evasion mechanism. J. Virol. 2008, 82, 9337–9344. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Seidel, C.A.; Cines, D.B. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 1984, 309, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.; Wang, L.; Mastellos, D.; Sahu, A.; Lambris, J.D.; Friedman, H.M. In vivo role of complement-interacting domains of herpes simplex virus type 1 glycoprotein gC. J. Exp. Med. 1999, 190, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, J.; Nagashunmugam, T.; Friedman, H.M. Viral interference with antibody and complement. Semin. Cell Dev. Biol. 1998, 9, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Salio, M.; Cella, M.; Suter, M.; Lanzavecchia, A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 1999, 29, 3245–3253. [Google Scholar] [CrossRef] [PubMed]
- Samady, L.; Costigliola, E.; MacCormac, L.; McGrath, Y.; Cleverley, S.; Lilley, C.E.; Smith, J.; Latchman, D.S.; Chain, B.; Coffin, R.S. Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (HSV) relieves the viral block to dendritic cell activation: potential of vhs- HSV vectors for dendritic cell-mediated immunotherapy. J. Virol. 2003, 77, 3768–3776. [Google Scholar] [CrossRef] [PubMed]
- Fruh, K.; Ahn, K.; Djaballah, H.; Sempe, P.; van Endert, P.M.; Tampe, R.; Peterson, P.A.; Yang, Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Jugovic, P.; York, I.; Russ, G.; Bennink, J.; Yewdell, J.; Ploegh, H.; Johnson, D. Herpes simplex virus turns off the TAP to evade host immunity. Nature 1995, 375, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Meyer, T.H.; Uebel, S.; Sempe, P.; Djaballah, H.; Yang, Y.; Peterson, P.A.; Fruh, K.; Tampe, R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996, 15, 3247–3255. [Google Scholar] [PubMed]
- Hill, A.B.; Barnett, B.C.; McMichael, A.J.; McGeoch, D.J. HLA class I molecules are not transported to the cell surface in cells infected with herpes simplex virus types 1 and 2. J. Immunol. 1994, 152, 2736–2741. [Google Scholar] [PubMed]
- Tigges, M.A.; Leng, S.; Johnson, D.C.; Burke, R.L. Human herpes simplex virus (HSV)-specific CD8+ CTL clones recognize HSV-2-infected fibroblasts after treatment with IFN-gamma or when virion host shutoff functions are disabled. J. Immunol. 1996, 156, 3901–3910. [Google Scholar] [PubMed]
- Trgovcich, J.; Johnson, D.; Roizman, B. Cell surface major histocompatibility complex class II proteins are regulated by the products of the gamma(1)34.5 and U(L)41 genes of herpes simplex virus 1 . J. Virol. 2002, 76, 6974–6986. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Eis-Hubinger, A.M.; Koch, N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J. Immunol. 2003, 171, 3075–3083. [Google Scholar] [PubMed]
- Stingley, S.W.; Ramirez, J.J.; Aguilar, S.A.; Simmen, K.; Sandri-Goldin, R.M.; Ghazal, P.; Wagner, E.K. Global analysis of herpes simplex virus type 1 transcription using an oligonucleotide-based DNA microarray. J. Virol. 2000, 74, 9916–9927. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Lee, S.Y.; Kim, S.Y.; Kim, J.K.; Kim, H.J.; Lee, H.M.; Choi, M.S.; Min, J.S.; Kim, M.J.; Choi, H.S.; Ahn, J.K. HSV-1 ICP27 suppresses NF-kappaB activity by stabilizing IkappaBalpha. FEBS Lett. 2008, 582, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; McLean, T.; Bachenheimer, S.L. Herpes Simplex Virus ICP27 Activation of Stress Kinases JNK and p38. J. Virol. 2005, 79, 8348–8360. [Google Scholar] [CrossRef] [PubMed]
- Gillis, P.A.; Okagaki, L.H.; Rice, S.A. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J. Virol. 2009, 83, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; Rice, S.; Bachenheimer, S.L. Herpes simplex virus type 1 ICP27-dependent activation of NF-kappaB. J. Virol. 2006, 80, 10565–10578. [Google Scholar] [CrossRef] [PubMed]
- Smiley, J.R. Herpes simplex virus virion host shutoff protein: immune evasion mediated by a viral RNase? J. Virol. 2004, 78, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Ahmed, C.M.; Dabelic, R.; Jager, L.D.; Noon-Song, E.N.; Haider, S.M.; Johnson, H.M.; Bigley, N.J. HSV-1-Induced SOCS-1 Expression in Keratinocytes: Use of a SOCS-1 Antagonist to Block a Novel Mechanism of Viral Immune Evasion . J. Immunol. 2009, . [Google Scholar]
- Mossman, K.L.; Macgregor, P.F.; Rozmus, J.J.; Goryachev, A.B.; Edwards, A.M.; Smiley, J.R. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 2001, 75, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Dahmus, M.E.; Rice, S.A. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J. Virol. 1997, 71, 2031–2040. [Google Scholar] [PubMed]
- Collins, S.E.; Noyce, R.S.; Mossman, K.L. Innate cellular response to virus particle entry requires IRF3 but not virus replication. J. Virol. 2004, 78, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Hardwicke, M.A.; Sandri-Goldin, R.M. The herpes simplex virus regulatory protein ICP27 contributes to the decrease in cellular mRNA levels during infection. J. Virol. 1994, 68, 4797–4810. [Google Scholar] [PubMed]
- McCarthy, A.M.; McMahan, L.; Schaffer, P.A. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 1989, 63, 18–27. [Google Scholar] [PubMed]
- Song, B.; Yeh, K.C.; Liu, J.; Knipe, D.M. Herpes simplex virus gene products required for viral inhibition of expression of G1-phase functions. Virology 2001, 290, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Knipe, D.M. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J. Virol. 2002, 76, 5893–5904. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Bruni, R.; Roizman, B. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 1997, 71, 1019–1024. [Google Scholar] [PubMed]
- Sin, J.I.; Kim, J.J.; Pachuk, C.; Satishchandran, C.; Weiner, D.B. DNA vaccines encoding interleukin-8 and RANTES enhance antigen-specific Th1-type CD4(+) T-cell-mediated protective immunity against herpes simplex virus type 2 In vivo. J. Virol. 2000, 74, 11173–11180. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Leib, D.A. Protection from primary infection and establishment of latency by vaccination with a herpes simplex virus type 1 recombinant deficient in the virion host shutoff (vhs) function. Vaccine 1998, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Geiss, B.J.; Smith, T.J.; Leib, D.A.; Morrison, L.A. Disruption of virion host shutoff activity improves the immunogenicity and protective capacity of a replication-incompetent herpes simplex virus type 1 vaccine strain. J. Virol. 2000, 74, 11137–11144. [Google Scholar] [CrossRef] [PubMed]
- Prechtel, A.T.; Turza, N.M.; Kobelt, D.J.; Eisemann, J.I.; Coffin, R.S.; McGrath, Y.; Hacker, C.; Ju, X.; Zenke, M.; Steinkasserer, A. Infection of mature dendritic cells with herpes simplex virus type 1 dramatically reduces lymphoid chemokine-mediated migration. J. Gen. Virol. 2005, 86, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Iwasaki, A. Autophagy and antiviral immunity. Curr. Opin. Immunol. 2008, 20, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Espert, L.; Codogno, P.; Biard-Piechaczyk, M. Involvement of autophagy in viral infections: antiviral function and subversion by viruses. J. Mol. Med. 2007, 85, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [PubMed]
- Alexander, D.E.; Leib, D.A. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 2008, 4, 101–103. [Google Scholar] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Melchjorsen, J.; Matikainen, S.; Paludan, S.R. Activation and Evasion of Innate Antiviral Immunity by Herpes Simplex Virus. Viruses 2009, 1, 737-759. https://doi.org/10.3390/v1030737
Melchjorsen J, Matikainen S, Paludan SR. Activation and Evasion of Innate Antiviral Immunity by Herpes Simplex Virus. Viruses. 2009; 1(3):737-759. https://doi.org/10.3390/v1030737
Chicago/Turabian StyleMelchjorsen, Jesper, Sampsa Matikainen, and Søren R. Paludan. 2009. "Activation and Evasion of Innate Antiviral Immunity by Herpes Simplex Virus" Viruses 1, no. 3: 737-759. https://doi.org/10.3390/v1030737
APA StyleMelchjorsen, J., Matikainen, S., & Paludan, S. R. (2009). Activation and Evasion of Innate Antiviral Immunity by Herpes Simplex Virus. Viruses, 1(3), 737-759. https://doi.org/10.3390/v1030737