Transcriptome and Small RNA Sequencing Analysis Revealed Roles of PaWB-Related miRNAs and Genes in Paulownia fortunei

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Transcriptome Sequencing and Data Analysis
2.3. Construction, Sequencing, and Identification of sRNAs
2.4. DERs Identification and Target Genes Prediction
2.5. Construction of the miRNA-Target Gene Interaction Network
2.6. qRT (Quantificational Real-Time Polymerase)-PCR Validation
3. Results
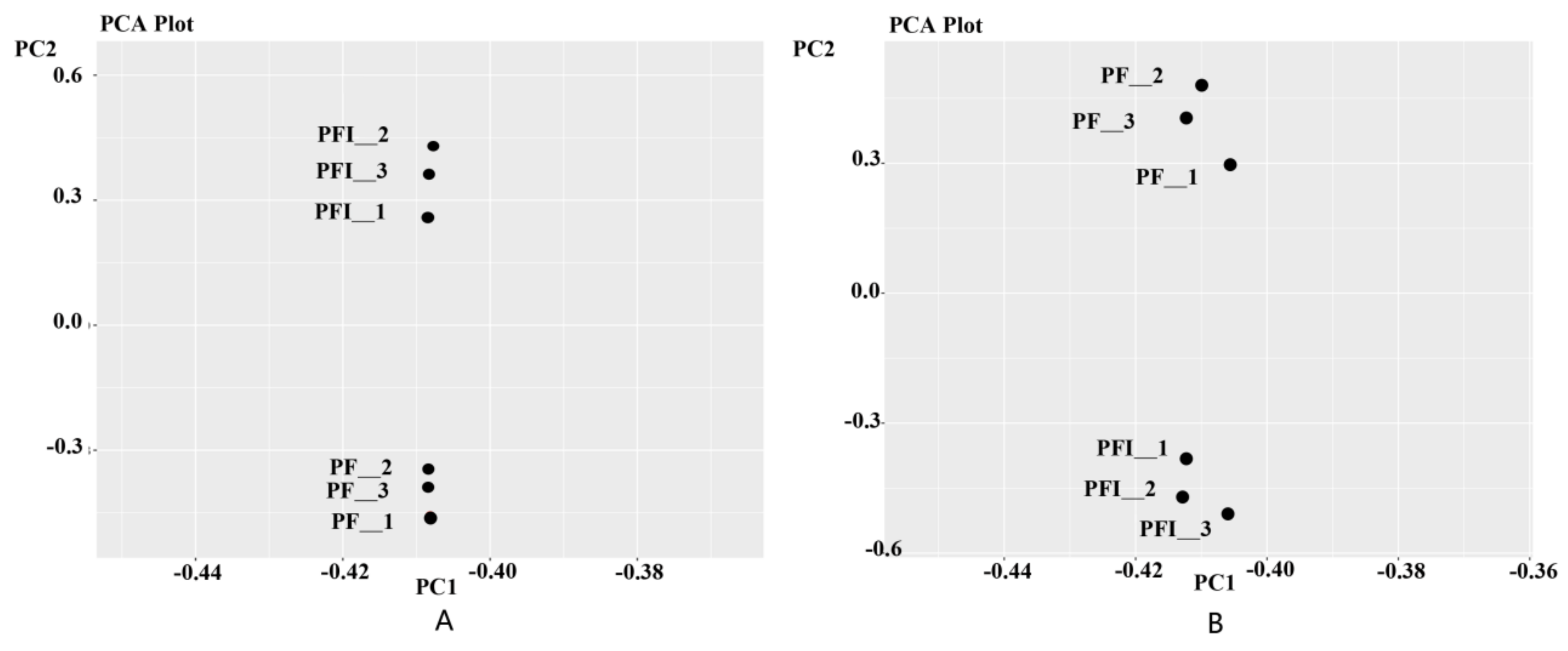
3.1. Transcriptome Sequencing Analysis
3.2. PaWB-Related Alternative Splicing (AS) Analysis
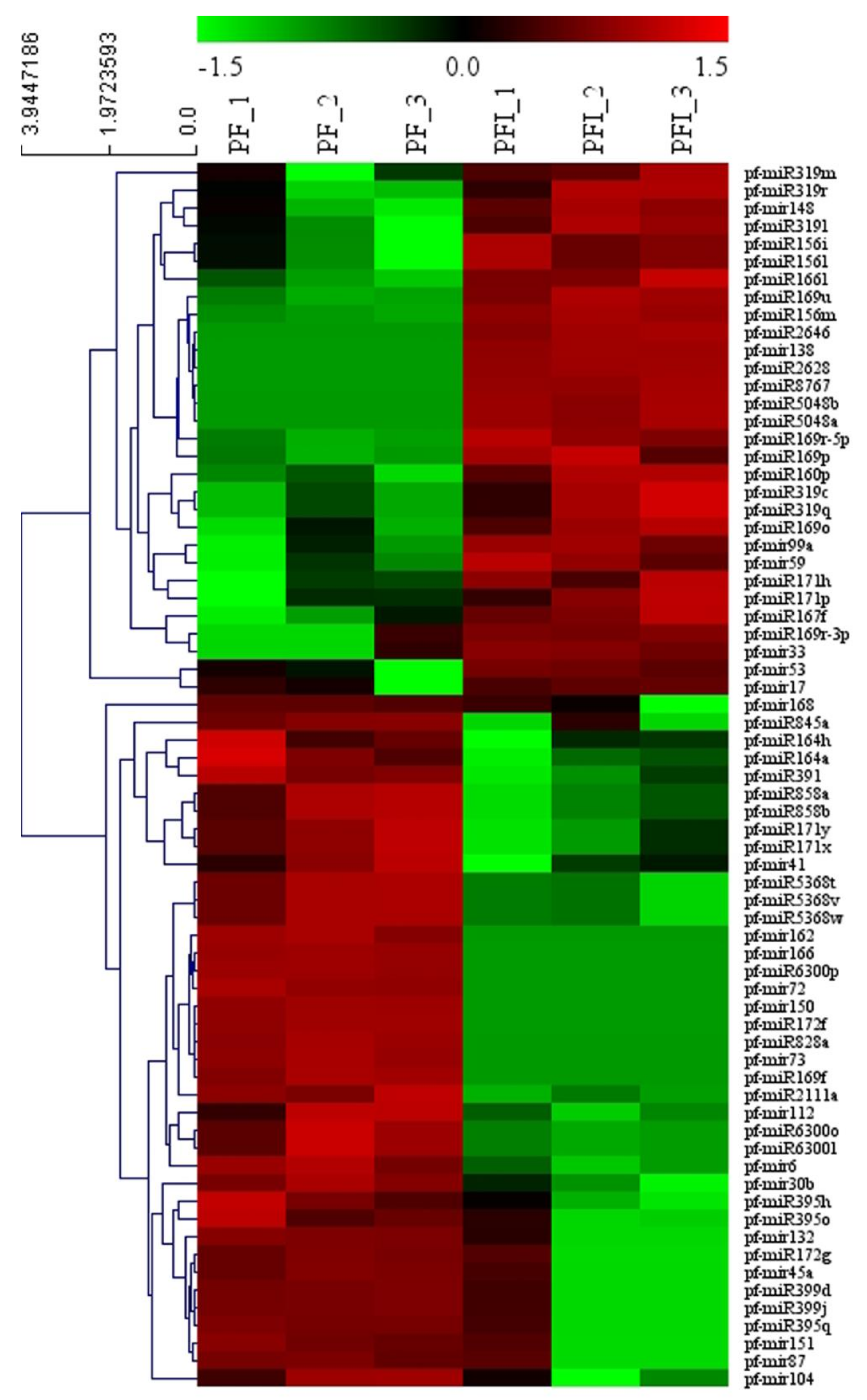
3.3. miRNA Identification
3.4. Functional Analysis of the PaWB-Related miRNAs and Their Target Genes
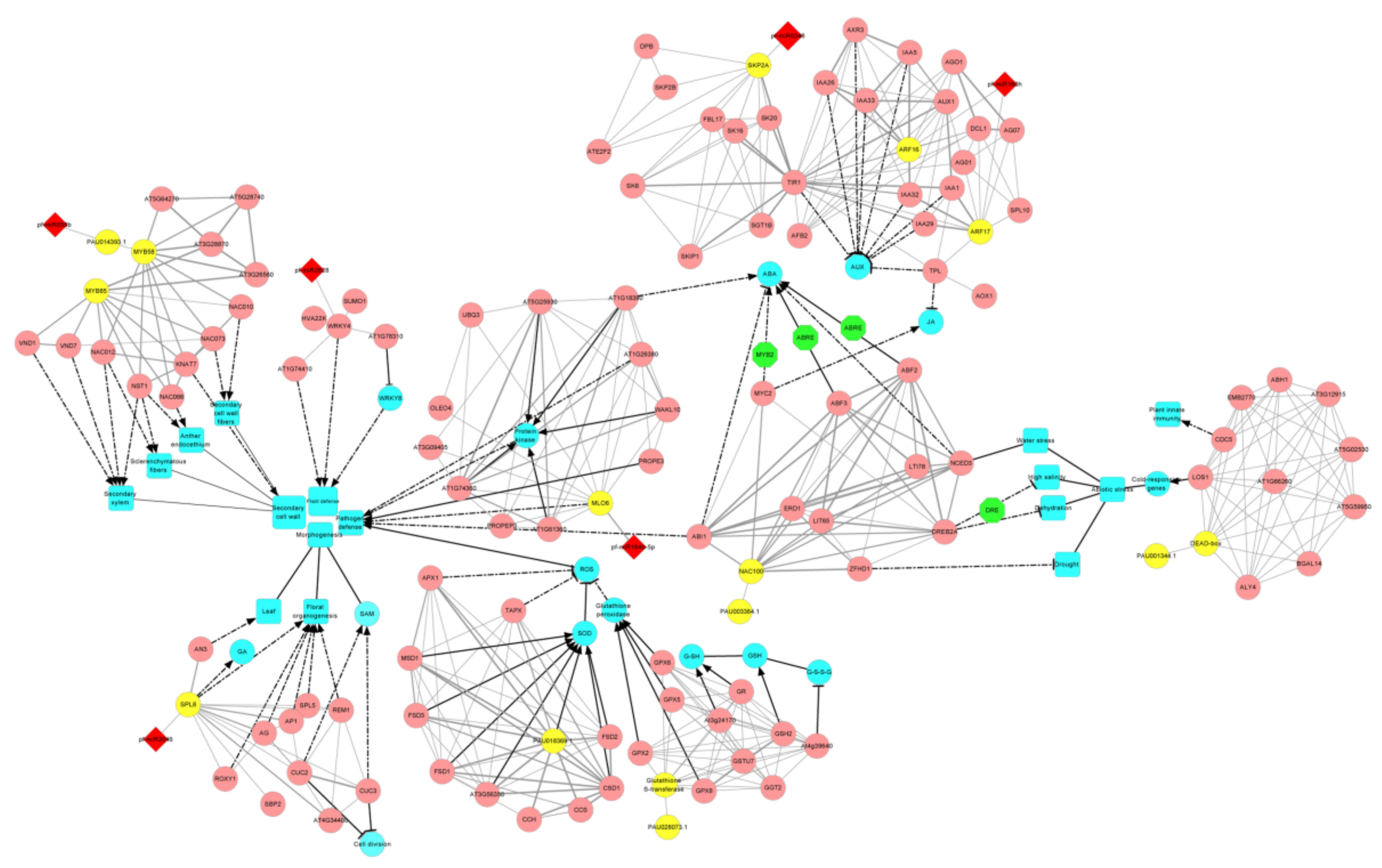
3.5. The miRNA-Target Gene Interaction Network That Related to PaWB
3.6. Verification of the Expression Patterns of miRNAs and Their Target Genes
4. Discussion
4.1. PaWB Responsive DER-Target Gene Pairs Regulate Morphological Variations
4.2. Regulation of Plant Defense Systems by DER-Target Genes Pairs
4.3. AS Events Related to PaWB Response in P. fortunei
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, B.; Zhao, Z.; Deng, M.; Cao, Y.; Dong, Y.; Fan, G. Effect of Salt Stress on Gene Expressions of Different Genotypes of Paulownia australis. J. Henan Agric. Univ. 2017, 4, 471–480. [Google Scholar]
- Jørgensen, I.; Vivekanandan, K. Private Forestry Based on Paulownia in Sri Lanka: An Appraisal of the Outgrower Scheme Presented by Paulownia Plantations LTD. Available online: http://www.diva-portal.org/smash/get/diva2:446489/FULLTEXT01.pdf (accessed on 3 July 2018).
- Ayan, S.; Sivacioglu, A.; Bilir, N. Growth variation of Paulownia Sieb. and Zucc. species and origins at the nursery stage in Kastamonu-Turkey. J. Environ. Biol. 2006, 27, 499–504. [Google Scholar] [PubMed]
- Ates, S.; Ni, Y.; Akgul, M.; Tozluoglu, A. Characterization and evaluation of Paulownia elongota as a raw material for paper production. Afr. J. Biotechnol. 2008, 7, 4153–4158. [Google Scholar]
- Hiruki, C.; Lemattre, M.; Lemattre, P.; Lemaire, F. Paulownia witches’-broom disease important in East Asia. Acta Hortic. 1999, 496, 63–68. [Google Scholar] [CrossRef]
- Shao, F.; Zhang, Q.; Liu, H.; Lu, S.; Qiu, D. Genome-wide identification and analysis of MicroRNAs involved in witches’-broom phytoplasma response in Ziziphus jujuba. PLoS ONE 2016, 11, e0166099. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Xu, E.; Deng, M.; Zhao, Z.; Niu, S. Phenylpropanoid metabolism, hormone biosynthesis and signal transduction-related genes play crucial roles in the resistance of Paulownia fortunei to paulownia witches’ broom phytoplasma infection. Genes Genom. 2015, 37, 913–929. [Google Scholar] [CrossRef]
- Jin, Q.; Wang, D.; Zhiqiang, L.; Wei, J.; Wang, Y.; Su, X. Molecular identification of phytoplasma strain isolated from mulberry trees infected by mulberry mosaic leaf roll disease in Zhejiang province. Sci. Seric. 2017, 42, 196–201. [Google Scholar]
- Chung, W.; Chen, L.; Lo, W.; Lin, C.; Kuo, C. Comparative analysis of the peanut witches’-broom phytoplasma genome reveals horizontal transfer of potential mobile units and effectors. PLoS ONE 2013, 8, e62770. [Google Scholar] [CrossRef] [PubMed]
- Padovan, A.; Firrao, G.; Schneider, B.; Gibb, K. Chromosome mapping of the sweet potato little leaf phytoplasma reveals genome heterogeneity within the phytoplasmas. Microbiology 2000, 146, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Teranaka, M.; Yora, K.; Asuyama, H. Mycoplasma- or PLT group-like microorganisms found in the phloem elements of plants infected with mulberry dwarf, potato witches’ broom, aster yellows, or paulownia witches’ broom. Jpn. J. Phytopathol. 1967, 33, 259–266. [Google Scholar] [CrossRef]
- Bertaccini, A.; Duduk, B.; Paltrinieri, S.; Contaldo, N. Phytoplasmas and phytoplasma diseases: A severe threat to agriculture. Am. J. Plant Sci. 2014, 5, 1763–1788. [Google Scholar] [CrossRef]
- Namba, S. Phytoplasmas: A century of pioneering research. J. Gen. Plant Pathol. 2011, 77, 345–349. [Google Scholar] [CrossRef]
- Ishiie, T.; Doi, Y.; Yora, K.; Asuyama, H. Suppressive effects of antibiotics of tetracycline group on symptom development of mulberry dwarf disease. Jpn. J. Phytopathol. 2009, 33, 267–275. [Google Scholar] [CrossRef]
- Oshima, K.; Kakizawa, S.; Nishigawa, H.; Jung, H.; Wei, W.; Suzuki, S.; Arashida, R.; Nakata, D.; Miyata, S.; Ugaki, M. Reductive evolution suggested from the complete genome sequence of a plant-pathogenic phytoplasma. Nat. Genet. 2004, 36, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Oshima, K.; Maejima, K.; Namba, S. Genomic and evolutionary aspects of phytoplasmas. Front. Microbiol. 2013, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Sugio, A.; Maclean, A.; Kingdom, H.; Grieve, V.; Manimekalai, R.; Hogenhout, S. Diverse targets of phytoplasma effectors: From plant development to defense against insects. Annu. Rev. Phytopathol. 2011, 49, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Maclean, A.; Sugio, A.; Makarova, O.; Findlay, K.; Grieve, V.; Tóth, R.; Nicolassen, M.; Hogenhout, S. Phytoplasma effector sap54 induces indeterminate leaf-like flower development in Arabidopsis plants. Plant Physiol. 2011, 157, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, Y.; Zhang, Z. Network analysis reveals a common host–pathogen interaction pattern in Arabidopsis immune responses. Front. Plant Sci. 2017, 8, 893. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.; Fan, G.; Deng, M.; Zhao, Z.; Xu, E.; Cao, L. Discovery of microRNAs and transcript targets related to witches’ broom disease in Paulownia fortunei by high-throughput sequencing and degradome approach. Mol. Genet. Genom. 2016, 291, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Niu, S.; Zhao, Z.; Deng, M.; Xu, E.; Wang, Y.; Yang, L. Identification of microRNAs and their targets in Paulownia fortunei plants free from phytoplasma pathogen after methyl methane sulfonate treatment. Biochimie 2016, 127, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Fan, G.; Dong, Y.; Zhao, Z.; Deng, M.; Wang, Z.; Liu, W. Proteome profiling of paulownia seedlings infected with Phytoplasma. Front. Plant Sci. 2017, 8, 342. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Lu, J.; Zhu, S.; Lin, C.; Tian, G.; Xu, X.; Zhao, W. Transcriptomic analysis of Paulownia infected by paulownia witches’-broom Phytoplasma. PLoS ONE 2013, 8, e77217. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhai, X.; Deng, M.; Fan, G. Relationship between Metabolites Variation and Paulownia Witches’ Broom. Sci. Silvae Sin. 2017, 6, 85–93. [Google Scholar]
- Wang, M.; Wang, Q.; Zhang, B. Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 2013, 530, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bao, M.; Sun, Y.; Yang, Y.; Xu, X.; Wang, J.; Han, N.; Bian, H.; Zhu, M. Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 2011, 77, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Kim, V. Microrna biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Dunoyer, P.; Jay, F.; Arnold, B.; Dharmasiri, N.; Estelle, M.; Voinnet, O.; Jones, J. A plant miRNA contributes to antibacterial resistance by repressing Auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.; Jay, F.; Nomura, K.; He, S.; Voinnet, O. Suppression of the microRNA pathway by bacterial effector proteins. Science 2008, 321, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Kasschau, K.; Xie, Z.; Allen, E.; Llave, C.; Chapman, E.; Krizan, K.; Carrington, J. P1/HC-Pro, a Viral suppressor of RNA silencing, interferes with Arabidopsis development and miRNA function. Dev. Cell 2003, 4, 205–217. [Google Scholar] [CrossRef]
- Abdi, H.; Williams, L. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Xu, E.; Fan, G.; Niu, S.; Zhao, Z.; Deng, M.; Dong, Y. Transcriptome sequencing and comparative analysis of diploid and autotetraploid Paulownia australis. Tree Genet. Genomes 2015, 11, 5. [Google Scholar] [CrossRef]
- Altschul, S.; Madden, T.; Schäffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar]
- Liu, H.; Zhao, Z.; Wang, L.; Deng, M.; Zhai, X.; Dong, Y.; Fan, G. Genome-wide expression analysis of transcripts, microRNAs, and the degradome in Paulownia tomentosa under drought stress. Tree Genet. Genomes 2017, 13, 128. [Google Scholar] [CrossRef]
- Hofacker, I.; Fontana, W.; Stadler, P.; Bonhoeffer, L.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte Chem. 1994, 125, 167–188. [Google Scholar]
- Meyers, M.; Chen, P.; Lin, Y.; Seki, Y. Biological materials: Structure and mechanical properties. Prog. Mater. Sci. 2008, 53, 1–206. [Google Scholar] [CrossRef]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J.; Zhuo, R. Integration of small RNAs, degradome and transcriptome sequencing in hyperaccumulator Sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016, 14, 1470–1483. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huertacepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K. String v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Duque, P. A role for SR proteins in plant stress responses. Plant Signal. Behav. 2011, 6, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Howell, M.; Kasschau, K.; Chapman, E.; Sullivan, C.; Cumbie, J.; Givan, S.; Law, T.; Grant, S.; Dangl, J. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of miRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Wei, H.; Wu, M.; Song, M.; Zhang, J.; Yu, J.; Fan, S.; Yu, S. Comparative expression profiling of miRNA during anther development in genetic male sterile and wild type cotton. BMC Plant Biol. 2013, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.; Oliver, P.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wen, L.; Zhu, H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci. 2015, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Fan, G.; Cao, X.; Niu, S.; Deng, M.; Zhao, Z.; Dong, Y. Transcriptome, microRNA, and degradome analyses of the gene expression of Paulownia with phytoplamsa. BMC Genom. 2015, 16, 896. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhai, X.; Zhang, Y.; Cheng, Z.; Li, X.; Fan, G. Comparative Analysis of MicroRNA Expression in Three Paulownia Species with Phytoplasma Infection. Forests 2018, 6, 302. [Google Scholar] [CrossRef]
- Gou, J.; Debnath, S.; Sun, L.; Flanagan, A.; Tang, Y.; Jiang, Q.; Wen, J.; Wang, Z. From model to crop: Functional characterization of SPL8 in M. truncatula led to genetic improvement of biomass yield and abiotic stress tolerance in alfalfa. Plant Biotechnol. J. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Unte, U.; Sorensen, A.; Pesaresi, P.; Gandikota, M.; Leister, D.; Saedler, H.; Huijser, P. SPL8, an SBP-box gene that affects pollen Sac development in Arabidopsis. Plant Cell 2003, 15, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Sun, T. Gibberellin-GID1-DELLA: A pivotal regulatory module for plant growth and development. Plant Physiol. 2010, 154, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Carol, P.; Richards, D.; King, K.; Cowling, R.; Murphy, G.; Harberd, N. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev. 1997, 11, 3194–3205. [Google Scholar] [CrossRef] [PubMed]
- Usami, T.; Horiguchi, G.; Yano, S.; Tsukaya, H. The more and smaller cells mutants of Arabidopsis thaliana identify novel roles for SQUAMOSA PROMOTER BINDING PROTEIN-LIKE genes in the control of heteroblasty. Development 2009, 136, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Song, Y.; Chen, Z.; Yu, D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, G.; Nakayama, H.; Ishikawa, N.; Kubo, M.; Demura, T.; Fukuda, H.; Tsukaya, H. ANGUSTIFOLIA3 plays roles in adaxial/abaxial patterning and growth in leaf morphogenesis. Plant Cell Physiol. 2011, 52, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Llorente, F.; Musksktt, P.; Sanchez-Vallet, P.; Lopez, G.; Ramos, B.; Sánchez-Rodríguez, C.; Jordá, L.; Parker, J.; Molina, A. Repression of the auxin response pathway increases Arabidopsis susceptibility to necrotrophic fungi. Mol. Plant 2008, 1, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Dowd, C.; Wilson, I.; Mcfadden, H. Gene expression profile changes in cotton root and hypocotyl tissues in response to infection with Fusarium oxysporum f. sp. Vasinfectum. Mol. Plant-Microbe Interact. 2004, 17, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Challa, G.; Mukesh, J. Role of auxin-responsive genes in biotic stress responses. Plant Signal. Behav. 2009, 4, 846–848. [Google Scholar]
- Fan, G.; Dong, Y.; Deng, M.; Zhao, Z.; Niu, S.; Xu, E. Plant-pathogen interaction, circadian rhythm, and hormone-related gene expression provide indicators of phytoplasma infection in Paulownia fortunei. Int. J. Mol. Sci. 2014, 15, 23141–23162. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, O.; Kochetov, A. Plant cell wall and mechanisms of resistance to pathogens. Russ. J. Genet. Appl. Res. 2016, 6, 622–631. [Google Scholar] [CrossRef]
- Zhong, R.; Demura, T.; Ye, Z. SND1, a NAC domain transcription factor, is a key regulator of secondary wall synthesis in fibers of Arabidopsis. Plant Cell 2006, 18, 3158–3170. [Google Scholar] [CrossRef] [PubMed]
- Deluc, L.; Barrieu, F.; Marchive, C.; Lauvergeat, V.; Decendit, A.; Richard, T.; Carde, J.P.; Mérillon, J.; Hamdi, S. Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 2006, 140, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Roy, S. Genome-wide sequential, evolutionary, organizational and expression analyses of phenylpropanoid biosynthesis associated MYB domain transcription factors in Arabidopsis. J. Biomol. Struct. Dyn. 2018, 36, 1577–1601. [Google Scholar] [CrossRef] [PubMed]
- Borevitz, J.; X, Y.; Blount, J.; Dixon, R.; Lamb, C. Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 2000, 12, 2383–2394. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Richardson, E.; Ye, Z. Two NAC domain transcription factors, SND1 and NST1, function redundantly in regulation of secondary wall synthesis in fibers of Arabidopsis. Planta 2007, 225, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Lee, C.; Zhou, J.; Mccarthy, R.; Ye, Z. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell 2008, 20, 2763–2782. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Tax, F. Receptor-like kinases: Key regulators of plant development and defense. J. Integr. Plant Biol. 2013, 55, 1184–1187. [Google Scholar]
- Shiu, S.; Bleecker, A. Plant receptor-like kinase gene family: Diversity, function, and signaling. Sci. STKE 2001, re22. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Singh, A.; Chand, R.; Singh, B. Genome wide analysis of disease resistance mlo gene family in sorghum [Sorghum Bicolor (l.) Moench]. Int. J. Plant Genom. 2012, 2, 18–27. [Google Scholar]
- Kemmerling, B.; Schwedt, A.; Rodriguez, P.; Mazzotta, S.; Frank, M.; Qamar, S.; Mengiste, T.; Betsuyaku, S.; Parker, J.; Müssig, C.; et al. The bri1-associated kinase 1, bak1, has a brassinolide-independent role in plant cell-death control. Curr. Biol. 2007, 17, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Huffaker, A. Endogenous peptide elicitors in higher plants. Curr. Opin. Plant Biol. 2011, 14, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhao, Y.; Walker, R.; Berkowitz, G. Molecular steps in the immune signaling pathway evoked by plant elicitor peptides: Ca2+-dependent protein kinases, nitric oxide, and reactive oxygen species are downstream from the early Ca2+ signal. Plant Physiol. 2013, 163, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, S.; Kim, J.; Chun, H.; Choi, M.; Chung, W.; Moon, B.; Kang, C.; Park, C.; Yoo, J.; et al. Mlo, a modulator of plant defense and cell death, is a novel calmodulin-binding protein. Isolation and characterization of a rice mlo homologue. J. Biol. Chem. 2002, 277, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Peterhänsel, C.; Schulze-Lefert, P. Interaction analyses of genes required for resistance responses to powdery mildew in barley reveal distinct pathways leading to leaf cell death. Plant Cell 1997, 9, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, H.; Dechert, C.; Kogel, K.; Hückelhoven, R. A small GTP-binding host protein is required for entry of powdery mildew fungus into epidermal cells of barley. Plant Physiol. 2002, 128, 1447–1454. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Miklis, M.; Schmelzer, E.; SchulzeLefert, P.; Panstruga, R. Recruitment and interaction dynamics of plant penetration resistance components in a plasma membrane microdomain. Proc. Natl. Acad. Sci. USA 2005, 102, 3135–3140. [Google Scholar] [CrossRef] [PubMed]
- Durner, J.; Shah, J.; Klessig, D. Salicylic acid and disease resistance in plants. Crit. Rev. Plant Sci. 1999, 18, 547–575. [Google Scholar] [CrossRef]
- Wang, X.; Gao, J.; Zhu, Z.; Dong, X.; Wang, X.; Ren, G.; Zhou, X.; Kuai, B. TCP transcription factors are critical for the coordinated regulation of ISOCHORISMATE SYNTHASE 1 expression in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2015, 82, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.; Lewandowska, D.; Fuller, J.; Maronova, M.; Kalyna, M.; Davidson, D.; McNicol, J.; Raczynska, D.; Jarmolowski, A.; Barta, A.; et al. Alternative splicing in plants. Biochem. Soc. Trans. 2008, 36, 508–510. [Google Scholar] [CrossRef] [PubMed]
- Laloum, T.; Martín, G.; Duque, P. Alternative splicing control of abiotic stress responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Whitham, S.; Dineshkumar, S.; Choi, D.; Hehl, R.; Corr, C.; Baker, B. The product of the tobacco mosaic virus resistance gene N: Similarity to toll and the interleukin-1 receptor. Cell 1994, 78, 1101–1115. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Z.; Jing, B.; Gannon, P.; Ding, J.; Xu, F.; Li, X.; Zhang, Y. Transportin-SR is required for proper splicing of Resistance genes and plant immunity. PLoS Genet. 2011, 7, e1002159. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Kirti, P. Novel role for a serine/arginine-rich splicing factor, AdRSZ21 in plant defense and HR-like cell death. Plant Mol. Biol. 2012, 80, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J. Stress-Induced Alternative Splicing of Serine/Arginine-Rich Proteins in the Moss Physcomitrella Patens. Available online: http://www.diva-portal.org/smash/get/diva2:446489/FULLTEXT01.pdf (accessed on 3 July 2018).
- Lopato, S.; Gattoni, R.; Fabini, G.; Stevenin, J.; Barta, A. A novel family of plant splicing factors with a Zn knuckle motif: Examination of RNA binding and splicing activities. Plant Mol. Biol. 1999, 39, 761–773. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Primer (5′-3′) | Reverse Primers (5′-3′) |
|---|---|---|
| pf-miR160h | TGCCTGGCTCCCTGTATGCCT | |
| pf-miR164b-5p | TGGAGAAGCAGGGCACGTGC | |
| pf-miR858b | TTCGTTGTCTGTTCGACCTTG | |
| pf-miR167h | TAAAGCTGCCAGCGTGATCTA | |
| pf-miR156p | GCTCTCTATTCTTCTGTCATC | |
| pf-miR5048 | AAAACCAAAAAAGTAATGA | |
| pf-miR169a-3p | TGGCTACAATTTGCTCTCTTC | |
| pf-miR172e | TGAATCTTGATGATGCTGCATAA | |
| PAU000742.1 | ATGGCTGGTGGAATTAGTA | AATCTGGATGCTGTGGTA |
| PAU003364.1 | CTTCAGCCATCTCCAATC | CAGTAATTCCACAAGCAATC |
| PAU027187.1 | CCATCAAGAACAAGAAACC | TTATTATTAGCCTCCTCTGC |
| PAU012184.1 | AGGAAGACAGGTGATGAT | CAGACTGGTTATGAATAGCA |
| PAU025602.1 | TATCCTCAAGCAGTCGTTA | CACTATACCTCAATCCATTCC |
| PAU012247.1 | CTTGATTCCACCAATGCTAT | ACCAGAACTCCATACACAT |
| PAU01444.1 | TGACAACTAATGAGACTGG | AAGAAGATAGGCTGAGAAG |
| PAU014627.1 | GGCACAGAGCATAACATT | TTCCAGAGATTCCAACTTC |
| PAU029039.1 | TAGCAAGTCTGACTCCAA | GCAGTAAACAAGAGCAAAG |
| PAU012221.1 | GACAAGCGATTAGGCATT | ACAGTTCCTCTATCCTTCTT |
| PAU018369.1 | GTATATCAACAGGAGCACAT | CCTCAGCATTAGCAACTAT |
| PAU023427.1 | GCTGTGTTAATTCAGTCAAG | CTTCGGTCAAGAACTTCA |
| PAU001344.1 | CTTGATGAGGCAGATAGAATG | CACCTTGAGGAGGCATAT |
| PAU012606.1 | GGAGGAGGCATTCTTAAC | CGGACCAGGAATAATAATCT |
| PAU028073.1 | TGATGCCTATGAGACTTG | AGGACTTGGATACACTCT |
| PAU002956.1 | GGTGGCTTGAAGTAGATG | GCTGAAGTCTGAAGAACA |
| PAU015980.1 | ATGTGGAGAGGAGGAATG | TGGTCACTGTTGATAGCA |
| PAU007073.2 | CATCACCATCCGTATTGT | GGCTTCACTCTATCCTTC |
| PAU022499.1 | AGTGTAGAGTGTGCTTGTC | CTTTGTGGAATGTGTGCTT |
| Sample | Total Raw Reads | Total Valid Reads | Q20 (%) | GC Content (%) |
|---|---|---|---|---|
| PF_1 | 136,371,054 | 119,367,224 | 99.86 | 46.00 |
| PF_2 | 133,864,544 | 123,717,278 | 99.53 | 45.50 |
| PF_3 | 125,474,542 | 113,284,088 | 99.67 | 45.00 |
| PFI_1 | 130,262,456 | 124,472,802 | 99.81 | 47.00 |
| PFI_2 | 155,719,594 | 147,092,878 | 99.61 | 45.00 |
| PFI_3 | 137,646,316 | 131,027,110 | 99.63 | 45.50 |
| Sample | Valid Reads | Mapped Reads (%) | Unique Mapped Reads (%) | Multi Mapped Reads (%) |
|---|---|---|---|---|
| PF_1 | 84,989,606 | 47,528,895 (55.92) | 34,900,121 (41.06) | 12,628,774 (14.86) |
| PF_2 | 83,415,504 | 45,070,813 (54.03) | 36,987,732 (44.34) | 8,083,081 (9.69) |
| PF_3 | 76,364,322 | 41,534,953 (54.39) | 33,118,690 (43.37) | 8,416,263 (11.02) |
| PFI_1 | 56,829,426 | 25,302,842 (44.52) | 17,283,734 (30.41) | 8,019,108 (14.11) |
| PFI_2 | 56,829,426 | 42,396,529 (44.43) | 35,484,885 (37.19) | 6,911,644 (7.24) |
| PFI_3 | 88,618,880 | 39,074,874 (44.09) | 30,740,288 (34.69) | 8,334,586 (9.40) |
| AS_Category | PF_1 (% *) | PF_2 (%) | PF_3 (%) | PFI_1 (%) | PFI_2 (%) | PFI_3 (%) |
|---|---|---|---|---|---|---|
| AE | 5879 (9.51) | 5929 (9.23) | 5795 (9.07) | 5168 (9.07) | 6071 (9.49) | 5939 (9.36) |
| IR | 4660 (7.54) | 4742 (7.38) | 4688 (7.34) | 4286 (7.52) | 4800 (7.50) | 4790 (7.55) |
| MIR | 950 (1.54) | 959 (1.49) | 950 (1.49) | 901 (1.58) | 952 (1.49) | 955 (1.51) |
| MSKIP | 1009 (1.63) | 989 (1.54) | 1004 (1.57) | 805 (1.41) | 1004 (1.57) | 1016 (1.60) |
| SKIP | 4346 (7.03) | 4310 (6.71) | 4330 (6.78) | 3572 (6.27) | 4406 (6.89) | 4364 (6.88) |
| TSS | 19,838 (32.10) | 20,936 (32.58) | 20,911 (32.73) | 18,832 (33.05) | 20,607 (32.21) | 20,417 (32.19) |
| TTS | 18,309 (29.63) | 19,434 (30.24) | 19,451 (30.45) | 17,621 (30.93) | 19,141 (29.92) | 18,979 (29.92) |
| XAE | 1625 (2.63) | 1649 (2.57) | 1623 (2.54) | 1372 (2.41) | 1670 (2.61) | 1640 (2.59) |
| XIR | 2090 (3.38) | 2139 (3.33) | 2091 (3.27) | 1850 (3.25) | 2133 (3.33) | 2146 (3.38) |
| XMIR | 408 (0.66) | 409 (0.64) | 412 (0.64) | 373 (0.65) | 422 (0.66) | 416 (0.66) |
| XMSKIP | 474 (0.77) | 460 (0.72) | 479 (0.75) | 376 (0.66) | 457 (0.71) | 478 (0.75) |
| XSKIP | 2207 (3.57) | 2313 (3.60) | 2153 (3.37) | 1819 (3.19) | 2310 (3.61) | 2296 (3.62) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Zhai, X.; Cao, Y.; Zhao, H.; Wang, Z.; Liu, H.; Fan, G. Transcriptome and Small RNA Sequencing Analysis Revealed Roles of PaWB-Related miRNAs and Genes in Paulownia fortunei. Forests 2018, 9, 397. https://doi.org/10.3390/f9070397
Li B, Zhai X, Cao Y, Zhao H, Wang Z, Liu H, Fan G. Transcriptome and Small RNA Sequencing Analysis Revealed Roles of PaWB-Related miRNAs and Genes in Paulownia fortunei. Forests. 2018; 9(7):397. https://doi.org/10.3390/f9070397
Chicago/Turabian StyleLi, Bingbing, Xiaoqiao Zhai, Yabing Cao, Hui Zhao, Zhe Wang, Haifang Liu, and Guoqiang Fan. 2018. "Transcriptome and Small RNA Sequencing Analysis Revealed Roles of PaWB-Related miRNAs and Genes in Paulownia fortunei" Forests 9, no. 7: 397. https://doi.org/10.3390/f9070397
APA StyleLi, B., Zhai, X., Cao, Y., Zhao, H., Wang, Z., Liu, H., & Fan, G. (2018). Transcriptome and Small RNA Sequencing Analysis Revealed Roles of PaWB-Related miRNAs and Genes in Paulownia fortunei. Forests, 9(7), 397. https://doi.org/10.3390/f9070397