The Transcriptomic Responses of Pinus massoniana to Drought Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material and Experimental Setup
2.2. Total RNA Isolation, Sequencing Library Preparation and Transcriptome Assembly
2.3. Gene Expression Quantification and Differential Expression Analysis
2.4. Functional Annotation and Enrichment Analysis
2.5. qRT-PCR (Quantitative Real-Time PCR) Validation
3. Results
3.1. Variations in Phenotypes during Drought Stress
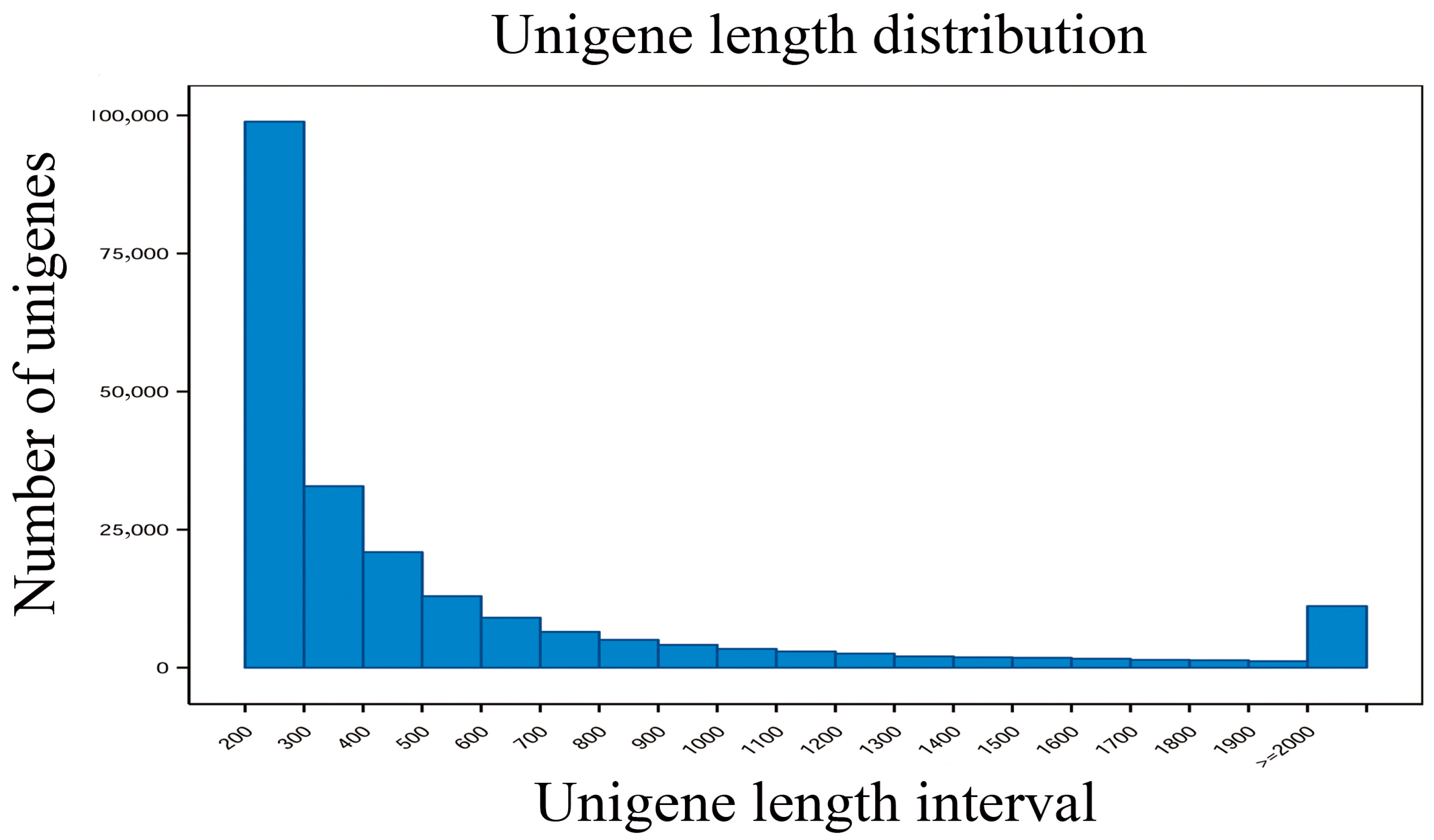
3.2. Transcriptome Sequencing and De Novo Assembly and Annotation
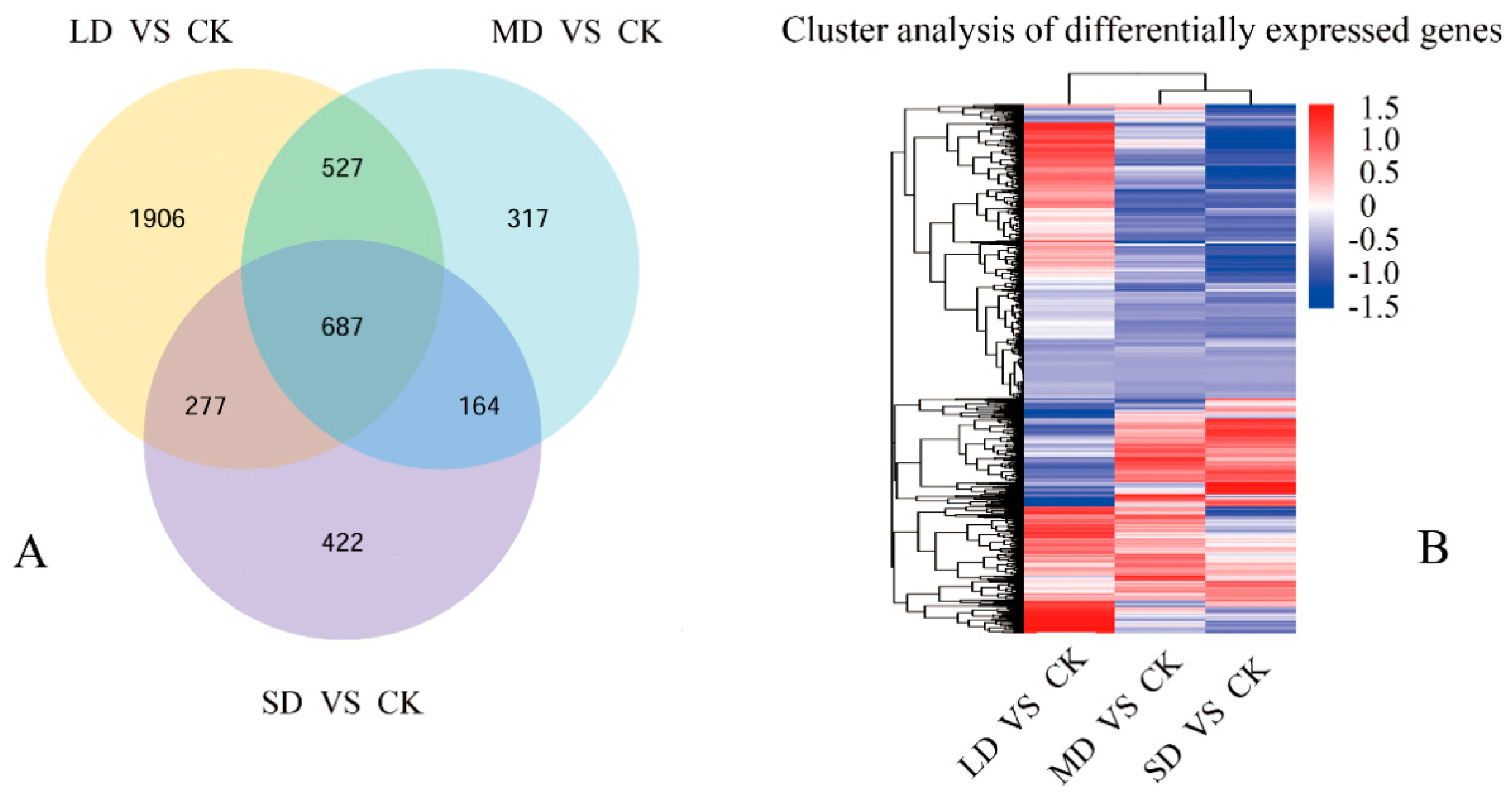
3.3. Exploration of Gene Expression in Seedlings under Drought Stress
3.4. Gene Ontology Enrichment Analysis
3.5. KEGG Pathway Enrichment Analysis
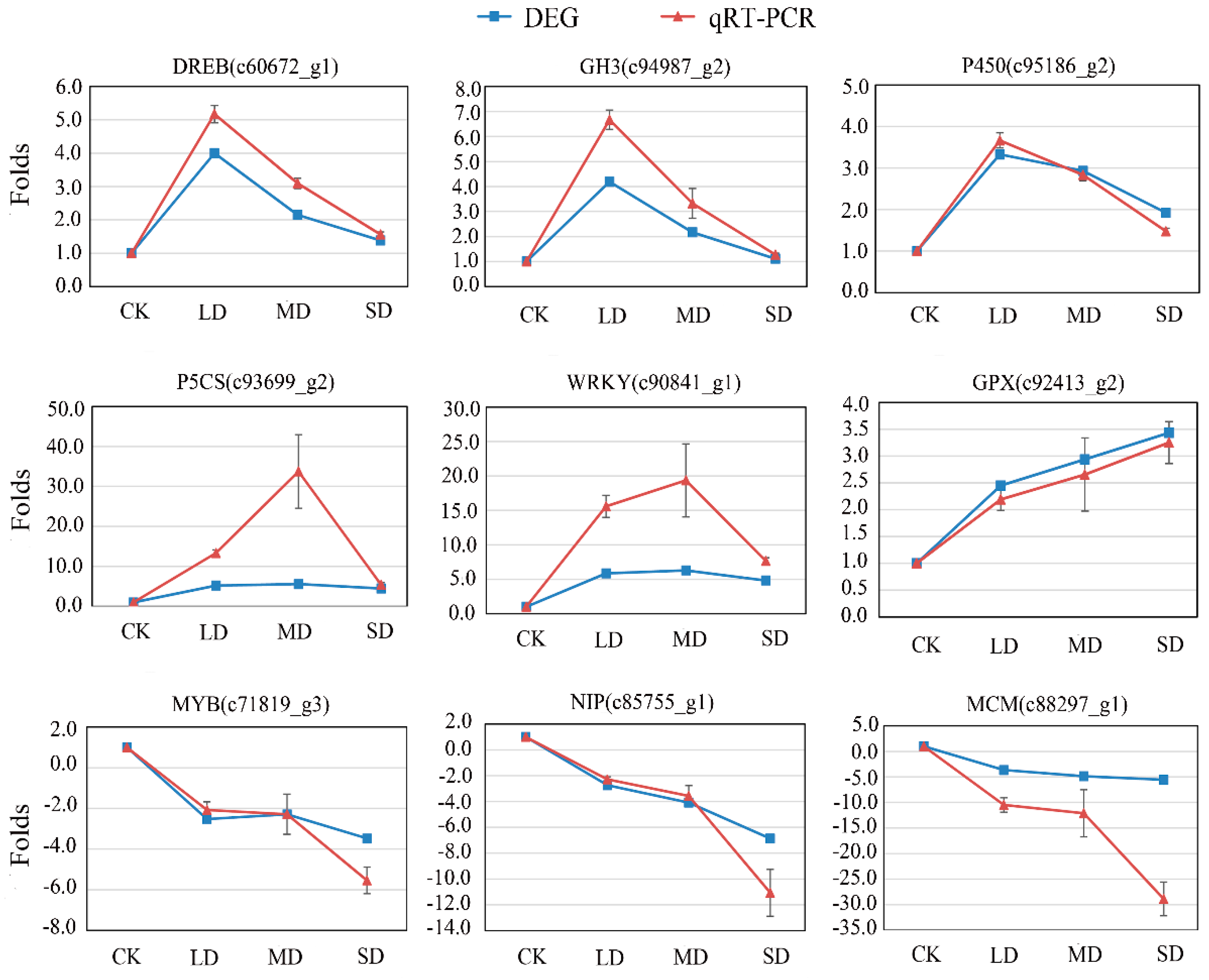
3.6. Validation by qRT-PCR
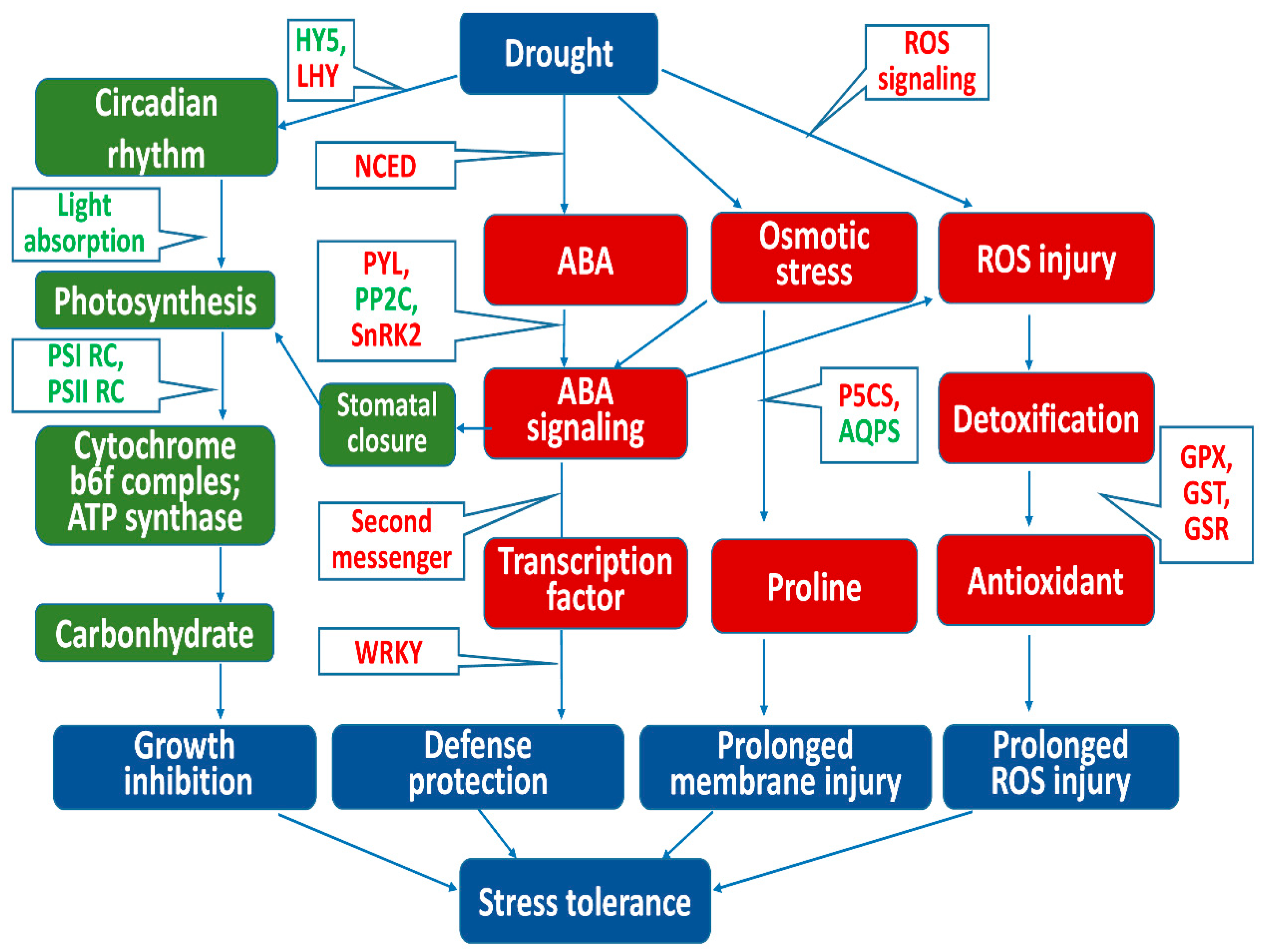
4. Discussion
4.1. Resistance to Osmotic Stress at Each Level of Drought Stress
4.2. Transcription Factors Responding to Stress Signals under Light Drought
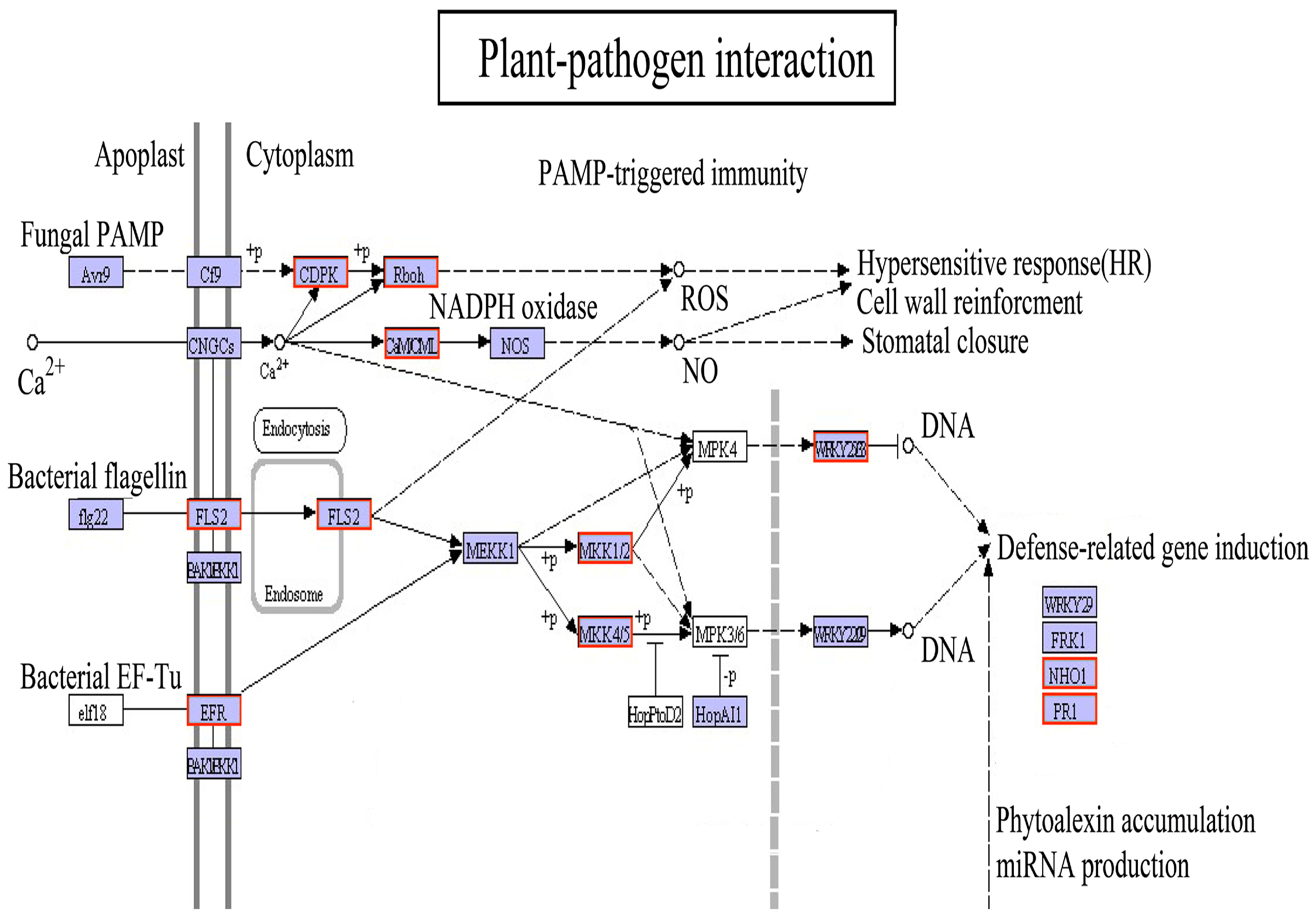
4.3. Defense Response of the Plant–Pathogen Interaction Pathway under Light Drought
4.4. ABA Response under Light and Moderate Drought Stress
4.5. Responses to Oxidative Stress under Moderate and Severe Drought Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, E.A. Plant responses to water deficit. Trends Plant Sci. 1997, 2, 48–54. [Google Scholar] [CrossRef]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Salo, H.M.; Sarjala, T.; Jokela, A.; Häggman, H.; Vuosku, J. Moderate stress responses and specific changes in polyamine metabolism characterize Scots pine somatic embryogenesis. Tree Physiol. 2016, 36, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, W.W.; Alba, R.; Yu, Y.-S.; Bordeaux, J.M.; Simões, M.; Dean, J.F.D. Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.). BMC Genom. 2011, 12, 264. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, P.; Soto, Á.; Collada, C. Comparative analysis of Pinus pinea and Pinus pinaster dehydrins under drought stress. Tree Genet. Genomes 2015, 11, 70. [Google Scholar] [CrossRef]
- Li, H.; Yao, W.; Fu, Y.; Li, S.; Guo, Q. De novo assembly and discovery of genes that are involved in drought tolerance in Tibetan Sophora moorcroftiana. PLoS ONE 2015, 10, e111054. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Zhou, Z.; Wang, Z. Cultivation and Utilization of Pulpwood Stand for Pinus massoniana; China Forestry Publishing House: Beijing, China, 2006. [Google Scholar]
- Du, M.; Ding, G.; Zhao, X. Responses to continuous drought stress and drought resistance comprehensive evaluation of different Masson pine families. Sci. Silvae Sin. 2017, 53, 21–29. [Google Scholar]
- Chen, H.; Yang, Z.; Hu, Y.; Tan, J.; Jia, J.; Xu, H.; Chen, X. Reference genes selection for quantitative gene expression studies in Pinus massoniana L. Trees 2016, 30, 685–696. [Google Scholar] [CrossRef]
- Cañas, R.A.; Feito, I.; Fuente-Maqueda, J.F.; Ávila, C.; Majada, J.; Cánovas, F.M. Transcriptome-wide analysis supports environmental adaptations of two Pinus pinaster populations from contrasting habitats. BMC Genom. 2015, 16, 909. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Ensminger, I.; Schmid, K.J. A catalogue of putative unique transcripts from Douglas-fir (Pseudotsuga menziesii) based on 454 transcriptome sequencing of genetically diverse, drought stressed seedlings. BMC Genom. 2012, 13, 673. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, G.J. Physiological responses of mycorrhizal Pinus massoniana seedlings to drought stress and drought resistance evaluation. Ying Yong Sheng Tai Xue Bao 2013, 24, 639–645. [Google Scholar] [PubMed]
- Ma, X.; Wang, P.; Zhou, S.; Sun, Y.; Liu, N.; Li, X.; Hou, Y. De novo transcriptome sequencing and comprehensive analysis of the drought-responsive genes in the desert plant Cynanchum komarovii. BMC Genom. 2015, 16, 753. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35 (Suppl. 2), W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Alicia, O. Gene ontology analysis for RNA-Seq: Accounting for selection bias. Gen. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Cui, B.; Zhang, T.; Qiao, G.; Ding, G.; Wen, X. The Temporal Transcriptomic Response of Pinus massoniana Seedlings to Phosphorus Deficiency. PLoS ONE 2014, 8, e105068. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Aroca, R.; Ferrante, A.; Vernieri, P.; Chrispeels, M.J. Drought, abscisic acid and transpiration rate effects on the regulation of PIP aquaporin gene expression and abundance in Phaseolus vulgaris plants. Ann. Bot. 2006, 98, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, G.; Gao, S.; Martinez, C.; He, G.; Zhou, Z.; Huang, X.; Lee, J.H.; Zhang, H.; Shen, Y.; et al. Arabidopsis transcription factor ELONGATED HYPOCOTYL5 plays a role in the feedback regulation of phytochrome a signaling. Plant Cell Online 2010, 22, 3634–3649. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, S.; Yamashino, T.; Nakanishi, H.; Mizuno, T. Functional characterization of HY5 homolog genes involved in early light-signaling in Physcomitrella patens. Biosci. Biotechnol. Biochem. 2011, 75, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.R.V.; Kumar, S.V.; Nandi, A.; Chattopadhyay, S. Functional interconnections of HY1 with MYC2 and HY5 in Arabidopsis seedling development. BMC Plant Biol. 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-R. Interaction between the Late Elongated hypocotyl (LHY) and Early flowering 3 (ELF3) genes in the Arabidopsis circadian clock. Genes Genom. 2012, 34, 329–337. [Google Scholar] [CrossRef]
- Wang, X.; Zeng, F.S.; Zhan, Y.G.; He, Z.L. Cloning and functional analysis of the circadian gene LHY promoter in fraxinus mandshurica. Plant Physiol. J. 2014, 50, 1675–1682. [Google Scholar] [CrossRef]
- Tang, W.; Charles, T.M.; Newton, R.J. Overexpression of the pepper transcription factor CaPF1 in transgenic virginia pine (Pinus Virginiana Mill.) confers multiple stress tolerance and enhances organ growth. Plant Mol. Biol. 2005, 59, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Guilfoyle, T.J.; Hagen, G. Auxin response factors. Curr. Opin. Plant Biol. 2007, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Liscum, E.; Reed, J.W. Genetics of Aux/IAA and ARF action in plant growth and development. Plant Mol. Biol. 2002, 49, 387–400. [Google Scholar] [CrossRef] [PubMed]
- De Diego, N.; Perez-Alfocea, F.; Cantero, E.; Lacuesta, M.; Moncalean, P. Physiological response to drought in radiata pine: Phytohormone implication at leaf level. Tree Physiol. 2012, 32, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Yin, L.; Qu, J.; Lu, J. Linkage of cold acclimation and disease resistance through plant-pathogen interaction pathway in Vitis amurensis grapevine. Funct. Integr. Genom. 2014, 14, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Bonello, P.; Gordon, T.R.; Herms, D.A.; Wood, D.L.; Erbilgin, N. Nature and ecological implications of pathogen-induced systemic resistance in conifers: A novel hypothesis. Physiol. Mol. Plant Pathol. 2006, 68, 95–104. [Google Scholar] [CrossRef]
- Kong, Q.; Qu, N.; Gao, M.; Zhang, Z.; Ding, X.; Yang, F.; Li, Y.; Dong, O.X.; Chen, S.; Li, X.; et al. The MEKK1-MKK1/MKK2-MPK4 kinase cascade negatively regulates immunity mediated by a mitogen-activated protein kinase kinase kinase in Arabidopsis. Plant Cell 2012, 24, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.T.; Dhekney, S.A.; Gray, D.J. PR-1 gene family of grapevine: a uniquely duplicated PR-1 gene from a Vitis interspecific hybrid confers high level resistance to bacterial disease in transgenic tobacco. Plant Cell Rep. 2011, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.S.; Kurjogi, M.M.; Khalil-Ur-Rehman, M.; Fiaz, M.; Pervaiz, T.; Jiu, S.T.; Jia, H.F.; Chen, W.; Fang, J.G. Grapevine immune signaling network in response to drought stress as revealed by transcriptomic analysis. Plant Physiol. Biochem. 2017, 121, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, J.; Chauhan, R.; Swarnkar, M.K.; Chahota, R.K.; Singh, A.K.; Shankar, R.; Yadav, S.K. Comprehensive transcriptomic study on horse gram (Macrotyloma uniflorum): De novo assembly, functional characterization and comparative analysis in relation to drought stress. BMC Genom. 2013, 14, 647. [Google Scholar] [CrossRef] [PubMed]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Jha, B. Transcription factors in plants and ABA dependent and independent abiotic stress signalling. Biol. Plant. 2010, 54, 201–212. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Xiong, L.; Li, W.; Zhu, J.-K.; Zhu, J. The plant cuticle is required for osmotic stress regulation of abscisic acid biosynthesis and osmotic stress tolerance in Arabidopsis. Plant Cell 2011, 23, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Ding, G. Dynamic of Volatiles and Endogenous Hormones in Pinus massoniana Needles under Drought Stress. Sci. Silvae Sin. 2017, 53, 49–54. [Google Scholar]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, X.; Song, J.; Zong, X.; Li, D.; Li, D. Over-expression of a Zea mays L. protein phosphatase 2C gene (ZmPP2C) in Arabidopsis thaliana decreases tolerance to salt and drought. J. Plant Physiol. 2009, 166, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mao, X.; Wang, C.; Jing, R. Overexpression of a common wheat gene TaSnRK2.8 enhances tolerance to drought, salt and low temperature in Arabidopsis. PLoS ONE 2010, 5, e16041. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-Q.; Chen, M.; Xu, Z.-S.; Zhao, C.-P.; Li, L.; Xu, H.-J.; Tang, Y.-M.; Zhao, X.; Ma, Y.-Z. The soybean GmbZIP1 transcription factor enhances multiple abiotic stress tolerances in transgenic plants. Plant Mol. Biol. 2011, 75, 537–553. [Google Scholar] [CrossRef] [PubMed]
- Dziri, S.; Hosni, K. Effects of cement dust on volatile oil constituents and antioxidative metabolism of Aleppo pine (Pinus halepensis) needles. Acta Physiol. Plant. 2012, 34, 1669–1678. [Google Scholar] [CrossRef]
- Zeng, Q.-Y.; Lu, H.; Wang, X.-R. Molecular characterization of a glutathione transferase from Pinus tabulaeformis (Pinaceae). Biochimie 2005, 87, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Gomez, L.; Vanacker, H.; Foyer, C.H. Interactions between biosynthesis, compartmentation and transport in the control of glutathione homeostasis and signalling. J. Exp. Bot. 2002, 53, 1283–1304. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.S.V.C.; Reddy, A.R. Glutathione reductase: A putative redox regulatory system in plant cells. In Sulfur Assimilation and Abiotic Stress in Plants; Khan, N.A., Singh, S., Umar, S., Eds.; Springer: Berlin, Germany, 2008; pp. 111–147. [Google Scholar]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, M.; Ding, G.; Cai, Q. The Transcriptomic Responses of Pinus massoniana to Drought Stress. Forests 2018, 9, 326. https://doi.org/10.3390/f9060326
Du M, Ding G, Cai Q. The Transcriptomic Responses of Pinus massoniana to Drought Stress. Forests. 2018; 9(6):326. https://doi.org/10.3390/f9060326
Chicago/Turabian StyleDu, Mingfeng, Guijie Ding, and Qiong Cai. 2018. "The Transcriptomic Responses of Pinus massoniana to Drought Stress" Forests 9, no. 6: 326. https://doi.org/10.3390/f9060326
APA StyleDu, M., Ding, G., & Cai, Q. (2018). The Transcriptomic Responses of Pinus massoniana to Drought Stress. Forests, 9(6), 326. https://doi.org/10.3390/f9060326