Abstract
DNA polymorphism at nine nuclear microsatellites of nine selected naturally-regenerated Norway spruce populations growing mainly within gene conservation units in different parts of the Czech Republic was studied. To verify the genetic quality of the selected gene conservation unit, we analyzed nine Norway spruce subpopulations from gene conservation unit GZ 102–Orlické hory. Genetic parameters can be used in state administrative decision making on including stands into gene conservation units. The level of genetic diversity within 17 investigated Czech Norway spruce units was relatively high. Mean values for the number of different alleles ranged from 12.2 (population SM 08) to 16.2 (subpopulation SM T4). The values of observed heterozygosity (Ho) ranged from 0.65 to 0.80 and expected heterozygosity (He) from 0.74 to 0.81. Pairwise population FST values ranging from 0.006 to 0.027 indicated low genetic differentiation between units, and values of Nei’s genetic distance among Norway spruce units ranged from 0.046 to 0.168, thus structuring of the investigated Norway spruce units was confirmed. Closer genetic similarity was seen in subpopulations from the gene conservation unit in Orlické hory than in the studied populations from other genetic conservation units. Additionally, the populations SM 01 and SM 05, both of Hurst ecotypes, were the closest to one another and the populations of mountain and alpine ecotypes were assembled into another group.
1. Introduction
Norway spruce (Picea abies (L.) Karsten) is a coniferous species belonging to the family Pinaceae. It is one of the most widespread tree species in Europe, where it is located mainly in Northern and Northeastern Europe and in the mountains of Central and Southern Europe. The natural distribution of this species within the territory of the Czech Republic is in the Hercyno-Carpathian region and Picea abies represents the only autochthonous species of the genus Picea growing in the Czech Republic. Picea abies belongs to the most variable taxon of its genus with a relatively large area of distribution [1]. There are three main important ecotypes in the Czech Republic differing morphologically and physiologically and growing at different altitudes. The alpine ecotype grows at an altitude above 1050 m. It is very resistant to wind, snow, and ice. Its crown is slim and dense, and the needles are stiff and short. The mountain ecotype grows at an altitude from 700 to 1050 m, and its crown is short and sparse. The Hurst ecotype grows at an altitude below 700 m. Its crown is broad and elliptical, and the needles are long, relatively sparse, and its cones are long [2]. Norway spruce is economically the most important tree species in the Czech Republic, where it is used for pulp and timber production. This species is very demanding of soil moisture and requires higher relative humidity. In the past 200 years, the spruce has been secondarily extended to everywhere in Central Europe, thereby pushing out most of the original trees [3]. Recently, the main coniferous area, such as that of spruce, pine, and larch, has gradually decreased in the Czech Republic, while the proportions of silver fir and deciduous trees have been increasing in order to achieve optimal species composition of forests [4]. A significant reduction today in the occurrence of spruce in some localities is due to the natural increase in drought caused by climate change [5]. One of the priority tasks under state forest policies and an international obligation of the Czech Republic is to conserve biodiversity in forest ecosystems while promoting the principles of sustainable management. Therefore, it is essential to determine the genetic variability in economically and ecologically valuable stands and populations. It is widely assumed that populations characterized by narrow genetic diversity could be more sensitive to environmental changes or disease, thereby leading to a decrease in productivity [6]. The genetic diversity of trees is crucial for the adaptation of forests to climate change [7,8] and for sustaining forest ecosystems [9]. Knowledge based on DNA analyses regarding the variability of genetic resources will contribute to the quality of the reproduction material and to creating an optimal species composition in forests. Norway spruce has been the subject of numerous genetic surveys using isozymes [10,11], expressed sequence tags markers [12,13], mitochondrial DNA [14,15], sequence tagged site markers [16,17], amplified fragment length polymorphism [18], single nucleotide polymorphisms [19,20], and microsatellite markers [21,22,23,24,25]. Nuclear simple sequence repeats (SSRs, or microsatellites) are widely used for assessing genetic diversity in forestry populations [6]. With their high degree of polymorphism, they provide an ideal tool for gene flow studies [26]. As co-dominant markers, they allow the assessment of heterozygosity. Nuclear SSR markers for Norway spruce have been developed from genomic dinucleotide and trinucleotide sequences and from expressed sequence tags (EST)-derived stretches [27]. The application of SSR markers developed from genomic DNA for Norway spruce is limited due to the tree’s large (ca. 20-gigabase) genome [28] and high proportion of repetitive DNA [21], which frequently produce complex multi-locus amplification products. To obtain a single locus, EST-SSRs markers derived from expressed regions have been developed [22,24]. These markers combine the advantages of microsatellite variability with the information content potentially carried by expressed sequences. Variable microsatellite markers relative to the coding regions are useful in forest population genetics (for example, in assessing adaptive variation) [22] and may be useful for association with phenotypic traits [24].
To provide insight into the levels of genetic variation and differentiation of selected naturally-regenerated Norway spruce populations and subpopulations growing mainly within the gene conservation units in different parts of the Czech Republic, eight EST-SSRs and one genomic-derived microsatellite markers were used. The gene conservation units are natural or man-made tree populations which are managed for maintaining evolutionary processes and adaptive potential across generations [29]. They present a set of forest stands with a significant share of valuable regional forest tree stocks in sufficient area to maintain the biological diversity of the population, which is able to reproduce on its own in case of suitable management methods. Each unit should have a designated status and a management plan, and one or more tree species recognized as target species for genetic conservation [29]. In the Czech Republic, the gene conservation units are proclaimed by the state administration in accordance with the valid legal regulation (Act No. 149/2003 Coll.) in order to save, conserve, and reproduce genetic resources of forest trees. According to Czech legislation, the gene conservation unit can be declared for one or more tree species in one or more separate parts and the size of one gene conservation unit should not be less than 100 ha. Natural regeneration should be preferred as a regeneration method. If artificial restoration is required, the reproductive material should originate from the same gene conservation unit.
Europe is an example of a complex where the distribution ranges of tree species extend across large geographical areas with profound environmental differences, and with ranges often overlapping across many countries. Conservation of forest genetic diversity through the use of gene conservation units is also conducted at the European level by European Forest Genetic Resources Programme (EUFORGEN), with which the Czech Republic is involved. This international cooperation supports the countries in their efforts to conserve forest genetic resources as part of sustainable forest management, as agreed in the context of Forest Europe, and contributes to developing genetic conservation strategies for forest trees at the pan-European level. Pan-European minimum requirements for dynamic conservation units of forest genetic diversity were developed as part of the EUFGIS project (Establishment of a European Information System on Forest Genetic Resources, April 2007–September 2010) which is one of the 17 actions co-funded by the European Commission through the Council Regulation (EC No 870/2004) on genetic resources in agriculture. Silvicultural interventions in gene conservation units should be allowed to enhance genetic processes, as needed, and field inventories carried out to monitor regeneration and the population size. These minimum requirements are now used by 36 countries to improve the management of forest genetic diversity.
Verification of the genetic quality of selected Czech genetic conservation units was carried out at the request of the state administration in order to develop procedures for acquiring knowledge about the genetic quality of other units, and these procedures will be used for subsequent monitoring strategies of the gene conservation units. Knowledge from genetic investigation is also important from the point of view of international cooperation (EUFORGEN) and it also contributes to fulfilling one of the priority tasks of the state forest policies and an international obligation of the Czech Republic to conserve biodiversity in forest ecosystems and promote the principles of sustainable management. In order to verify genetic quality of the selected gene conservation unit, we analyzed nine Norway spruce subpopulations from gene conservation unit GZ 102–Orlické hory. The results relating to genetic parameters can be used for state administrative decision making as to which stands to include into a gene conservation unit.
2. Materials and Methods
Eight Czech Norway spruce populations and nine subpopulations from the Orlické hory population (a total of 17 units) were genetically screened by nuclear microsatellites. Sampling of young needles was carried out during 2012–2016 from individual trees growing in gene conservation units, national parks, a protected landscape area, and national nature reserves. A more detailed investigation was carried out in the Orlické hory locality, where genetic parameters were studied within nine subpopulations of one gene conservation unit (GZ 102 Trčkov–Šerlišský kotel–Vrchmezí) and compared to the other populations. The locality designations, geographic coordinates, altitudes, and natural origins are presented in Table 1, sampling locations in Figure 1, and mean sample size of studied units was 34.7. The distance between randomly-sampled adult trees was approximately 80–100 m within gene conservation units in different parts of the Czech Republic, and 25–30 m within nine subpopulations of one gene conservation unit (GZ 102 Trčkov–Šerlišský kotel–Vrchmezí). Investigated Norway spruce populations or subpopulations represent three main different ecotypes (Table 1).

Table 1.
Geographic coordinates of the Picea abies units.
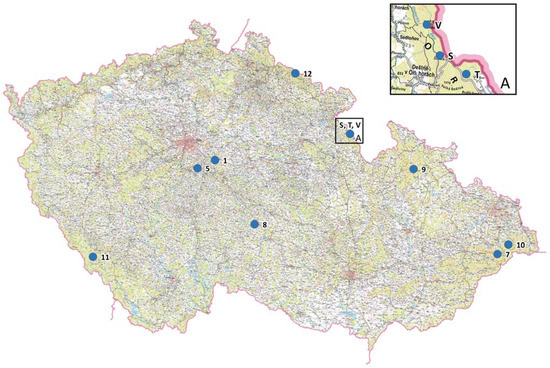
Figure 1.
Geographical locations of the Picea abies units.
Total genomic DNA was extracted from 20 mg of dry or 100 mg of frozen young needles collected from 591 Picea abies individuals of the investigated localities using a DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) while following the manufacturer’s instructions. Liquid nitrogen was used for disrupting the plant material. DNA concentrations and purity were measured spectrophotometrically using a NanoPhotometer (Implen, Munich, Germany). SSR markers were used as molecular genetic markers for detecting genetic variation in the selected Norway spruce units. The following markers with dinucleotide motifs, whose PCR products provided single, clear, reproducible patterns were used: PAAC23, PAAC19 [22], WS00716.F13, WS0092.A19, WS0022.B15, WS0073.H08, WS00111.K13, WS0023.B03 [24] derived from expressed sequence tags (EST-SSRs), and SpAGD1 [23] developed from genomic libraries of Norway spruce [21]. The nine nuclear microsatellite markers were assembled into three multiplexes from the viewpoint of the targeted allele sizes and amplification conditions. Specific primers were labelled fluorescently using FAM, VIC, and NED dyes. The amplification reaction conditions of the first multiplex with loci PAAC23, PAAC19, and SpAGD1 were for each sample in a final volume of 15 µL and contained 1 µL of template DNA (≈10–50 ng/µL), 1.5 µL of 10× PCR buffer (Mg-free), 0.2 mM of dNTP mixture (Takara Bio Inc., Otsu, Shiga, Japan), 2 mM MgCl2, 0.37 U of Platinum® Taq DNA polymerase (Invitrogen, Carlsbad, CA, USA), and primer combinations of the forward and reverse primers. The primer concentrations of loci PAAC19 and SpAGD1 were 0.1 µM and for locus PAAC23 0.2 µM. The reaction mixtures were supplemented with sterile water for molecular biology (Sigma-Aldrich, St. Louis, MO, USA). The PCR profile was as follows: initial denaturation at 94 °C for 3 min followed by 37 cycles of denaturation at 94 °C for 45 s, an annealing temperature of 57 °C for 45 s, and extension at 72 °C for 45 s, with a final extension step at 72 °C for 20 min. PCR for the second multiplex with loci WS00716.F13, WS0092.A19, and WS0022.B15 was performed using the Type-it® Microsatellite PCR Kit (Qiagen, Hilden, Germany). The concentration of each primer was 0.1 µM. PCR cycling conditions consisted of an initial denaturation at 95 °C for 15 min followed by 26 cycles of denaturation at 94 °C for 30 s, an annealing temperature at 53 °C for 90 s and extension at 72 °C for 30 s, with a final extension step at 60 °C for 30 min. PCR conditions for the third multiplex with loci WS0073.H08, WS00111.K13, and WS0023.B03 were the same as for the second multiplex, again with concentrations of each primer at 0.1 µM, except that the annealing temperature was 55 °C. Amplifications were carried out in a Veriti thermal cycler (Applied Biosystems, Foster City, CA, USA).
PCR products were separated by capillary electrophoresis using the Applied Biosystems 3500 genetic analyzer (Applied Biosystems, Foster City, CA, USA). As size standard, we used GeneScanTM 600LIZ® (Applied Biosystems, Foster City, CA, USA). Alleles were sized using GeneMapper® 4.1 software (Applied Biosystems, Foster City, CA, USA). Micro-Checker software was used for identifying and correcting genotyping errors in microsatellite data and for estimation of null allele frequencies [30]. The majority of genetic diversity parameters—number of alleles, Shannon’s information index, observed heterozygosity, expected heterozygosity, fixation index (F), pairwise population FST values of the genetic divergence, Nei’s genetic distance, and a principal coordinate analysis (PCoA) were calculated using the statistical program GenAlEx 6.501 [31,32]. The fixation index was calculated as F = 1 − (Ho/He) according to Wright [33]. Deviations from Hardy-Weinberg equilibrium (HWE) for studied loci were assessed using CERVUS 3.0.7 with Bonferroni correction for evaluating the deviation significance [34]. Global Hardy-Weinberg tests across the studied populations were performed by GENEPOP 4.2 [35,36] using Markov chain Monte Carlo simulations with 10,000 dememorizations, 100 batches, and 10,000 iterations to detect significant heterozygote excess or deficiency. The genetic divergence FST and Nei’s genetic distance [37] between populations were estimated by computing a pairwise population matrix. Total FST was calculated as the analysis of molecular variance (AMOVA) based on 999 permutations with the software GenAlEx 6.501 [31,32]. A chi-squared test was used for evaluating the Hardy–Weinberg equilibrium (HWE) for co-dominant genotypes at a single locus and for a single population. The number of private alleles is the number of alleles unique to a single population. It was calculated as the mean value from all studied loci. The Bayesian clustering method implemented in STRUCTURE 2.3.4 software [38,39,40,41] was used to infer the population structure. The admixture model and correlated allele frequencies were used. We used a Length of Burn-in Period of 10,000 and 100,000 Markov chain Monte Carlo (MCMC) Repeats after Burn-in. Multiple runs were performed by setting the number of populations (K) from K = 1 to K = 15. Each run was repeated ten times. The best estimate of K values was calculated using the web-based STRUCTURE HARVESTER program [42].
3. Results
Genetic diversity parameters were studied in the 17 Czech units (populations, subpopulations) using nine nuclear microsatellite markers. The nine subpopulations were taken from just one gene conservation unit (GZ 102 Trčkov–Šerlišský kotel–Vrchmezí) in order to compare the genetic distances among them and with the other studied populations situated in different localities of the Czech Republic.
The selected markers that generated simple patterns detected 23 (PAAC23), 36 (PAAC19), 38 (SPAGD1), 25 (WS00716.F13), seven (WS0092.A19), 24 (WS0022.B15), nine (WS0073.H08), 34 (WS00111.K13), and 37 (WS0023.B03) different alleles over 591 tested Norway spruce trees (Table 2). All SSR markers had dinucleotide repeats and their PCR products provided variable expected sizes. With one exception in subpopulation SM S4, the Micro-Checker software [30] found no evidence at any locus of scoring error due to stuttering and no evidence of large allele dropout for any unit. In SM S4, stuttering at locus PAAC23 might have resulted in scoring errors, as was indicated by the highly significant shortage of heterozygote genotypes with alleles of one repeat unit difference. Analyses indicated homozygote excess at loci PAAC19 and SpAGD1 in all units; at locus WS00716.F13 in units SM S1, SM V2, SM 05, and SM 11; at locus WS0022.B15 in units SM S1, SM S4, SM T1, and SM V2; at locus PAAC23 in subpopulation SM S4; at locus WS00111.K13 in subpopulation SM T1; at locus WS0073.H08 in population SM 01; and at locus WS0092.A19 in population SM 08. The Micro-Checker software indicated that these units are possibly in HWE while showing signs of a null allele. The evaluations of null allele frequencies in accordance with Oosterhout [30] are recorded in Table 2. Some significant deviations (p < 0.001) from HWE based on the chi-square test for HWE [31,32] were detected at locus PAAC23 in two units (SM T4, SM 05), at locus PAAC19 in 14 units (all except units SM V4, SM 01, and SM 12), at locus SpAGD1 (all except population SM 11), at locus WS0092.A19 (with significant deviation occurring in units SM T2, SM T4, SM V1, SM 01, and SM 08), at loci WS00716.F13 and WS0023.B03 (with significant deviation occurring only in population SM 05), at locus WS0022.B15 (in the SM S4 and SM V2 subpopulations), and at locus WS0073.H08 (in population SM 01). Significant deviations were not observed at locus WS00111.K13.
Table 2.
Characteristics of selected nuclear microsatellite loci across 17 investigated Norway spruce units.
The genetic diversity parameters with the primer sequences of the studied markers are reported in Table 2. There were 234 different alleles detected at the nine loci in the 591 Norway spruce individuals. Expected heterozygosity (He) ranged from 0.20 (WS0092.A19) to 0.93 (SpAGD1, WS00111.K13), with a mean value of 0.78. The mean value of observed heterozygosity (Ho) was 0.71 and ranged from 0.22 (WS0092.A19) to 0.97 (WS0023.B03). Shannon’s information index calculated for allelic and genetic diversity also depends on the evenness of allele frequencies, which ranged from 0.41 at locus WS0092.A19 to 2.86 at locus SpAGD1. Fixation index values varied from −0.075 (WS0023.B03) to 0.41 (PAAC19). Most of the loci exhibited homozygote excess with positive F values. The loci WS0073.H08, WS00111.K13, and WS0023.B03 with negative fixation indices reflect excesses of heterozygotes in comparison to their expected frequencies. According to the program CERVUS 3.0.7, significant deviations from HWE (p < 0.001) were detected at three loci (PAAC19; SpAGD1—deficiency of heterozygotes; WS0023.B03—excess of heterozygotes) across 17 investigated Norway spruce units.
Genetic diversity characteristics of the 17 investigated Norway spruce units are given in Table 3. The mean values of the different alleles ranged from 12.2 (SM 08) to 16.2 (SM T4). The highest number of different alleles, at 28, occurred in subpopulation SM T4 at locus SpAGD1. The lowest, at only two alleles, was observed in the subpopulations SM V1 and SM V4 at locus WS0092.A19. The mean effective number of alleles ranged from 6.5 (SM 08) to 9.9 (SM T4). Mean values of population genetic diversity according to Shannon’s information index (I) ranged from 1.93 to 2.19. The values of observed heterozygosity (Ho) ranged from 0.65 to 0.80 and of expected heterozygosity (He) from 0.74 to 0.81. Slightly negative fixation index (F) values occurred in two populations (SM 09 and SM 12). Other Norway spruce units showed positive values of fixation index (0.011–0.199), indicating heterozygote deficiencies relative to the expected fraction. Global Hardy-Weinberg test detected high significant deviations from HWE for all 17 Norway spruce units. The result “highly significant” is reported when at least one of the individual tests being combined yielded a zero p-value estimation.
Table 3.
Mean values for genetic characteristics of 17 investigated Norway spruce units from nine selected nuclear microsatellite loci.
The highest number (six) of private alleles was found in population SM 01, where one allele appeared at loci WS00716.F13, WS0073.H08, and WS00111.K13, and three alleles at locus WS0092.A19. In many units (SM V1, SM V2, SM T2, SM S4, SM T4, and SM 12), there were no private alleles. Significant allelic frequencies differences were found across the 17 studied units at most loci. For example, a 280 bp allele at locus PAAC23 varied in frequencies from 34% to 60%, a 165 bp allele at locus PAAC19 varied from 0% to 26%, and a 196 bp allele at WS0022.B15 from 1% to 17%.
Estimates of differentiation expressed by FST values ranging from 0.006 to 0.027 indicated low genetic differentiation between units. Differentiations between the nine subpopulations from the gene conservation unit GZ 102 Trčkov–Šerlišský kotel–Vrchmezí showed lower values (0.006–0.011). FST values were greater than zero and so confirm the structuring of the studied Norway spruce units. After calculation of total FST based on 999 permutations, the overall level of genetic differentiation was found to be very low (0.011). Genetic distances between units were calculated based on Nei’s standard genetic distance [37]. The longest Nei’s genetic distance (0.168) appeared between the SM S4 (Orlické hory–Šerlich) and SM 08 (Českomoravská vrchovina) units. The closest Nei’s genetic distance (0.046) was between the SM S2 and SM T2 subpopulations, both from the Orlické hory locality. Values of Nei’s genetic distance among subpopulations of the gene conservation unit ranged from 0.046 to 0.092. The closest pairwise Nei’s genetic distance within other observed localities was 0.062 between SM 01 (Středočeská pahorkatina) and SM 05 (Středočeská pahorkatina), both of the Hurst ecotype. Nei’s standard genetic distances among the units are graphically illustrated in Figure 2, constructed on the basis of principal coordinate analysis, which shows the genetic similarity of the subpopulations from the gene conservation unit GZ 102 Trčkov–Šerlišský kotel–Vrchmezí. An interesting result was the grouping of the same ecotypes (Hurst, alpine, and mountain). The spatial differentiation was not confirmed by the STRUCTURE analysis. The Bayesian analysis identified K = 2 as the most relevant number of clusters. The admixture of clusters was very similar for studied Norway spruce units.
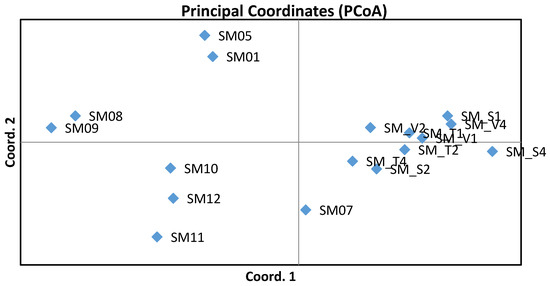
Figure 2.
Results of the principal component analysis.
4. Discussion
The aim of this work was to determine the genetic parameters of selected Norway spruce units growing in different parts of the Czech Republic, where their regeneration is a naturally ongoing process, and to compare the genetic similarities of nine subpopulations from one gene conservation unit (GZ 102—Orlické hory) to other populations distributed across the Czech Republic. Nuclear microsatellite markers were tested in genetic studies of spruce populations. Nine nuclear microsatellite loci were chosen with clear, reproducible PCR products of expected sizes and sufficient polymorphism.
The highest variability was found in locus SpAGD1 developed from genomic DNA. This was in accordance with Rungis et al. [24], who had found that the EST-SSRs showed significantly less variation than did the genomic-derived SSRs, as was the result also in his study, where the 25 EST-SSRs had approximately 9% less heterozygosity than did the 17 genomic-derived SSRs. However, the EST-SSR markers associated with the coding regions of the adaptive part of the spruce genome may be more useful in investigating defense against stress. At present, threats to this species are increasing at many localities of the Czech Republic, in particular due to climate changes and increasing water deficiency. It is generally presumed that populations with greater genetic diversity will have a stronger ability to adapt to changes in environmental conditions. Evaluation of genetic variation in genes controlling adaptive traits using genetic markers [8,13,20,43] could contribute to conserving the stability of long-lived forest trees. Analyzed Czech Norway spruce units have shown high levels of genetic diversity, so it can be said that these populations are appropriately included in the gene conservation unit. A prerequisite for these forests is their higher ability to adapt to changing environmental conditions. This knowledge is very important for forest management from the standpoint of utilizing the analyzed populations as important forest genetic resources of reproductive materials and for establishing genetic conservation strategies of gene conservation units. An interesting result was the grouping of studied populations according to three different ecotypes, as is illustrated by PCoA analysis. Already in a previous study of our workplace, similar results were found using six nuclear microsatellite loci, when populations from the České Švýcarsko National Park and the locality of Křivoklátsko, both Hurst ecotype, were distinguished from populations of mountain ecotype from locations Fláje (Krušné hory) and Trčkov–Šerlišský kotel (Orlické hory) based on PCoA analysis as well.
When we compared data based on nuclear microsatellites, we saw that the level of genetic diversity within the 17 investigated Czech Norway spruce units was relatively high and comparable with population diversities in such other countries as Austria [25], Poland [44], Italy [27], and Germany [45]. Diversity in Bosnia and Herzegovina populations was slightly lower in terms of the mean values of different alleles per population. Whereas in Czech populations, this ranged from 12.2 to 16.2, in Bosnia and Herzegovina populations, these means ranged from 7.8 to 15 [46]. The mean number of alleles at the same five loci (WS00716.F13, WS0022.B15, WS0073.H08, WS00111.K13, and WS0023.B03) over three Tyrol populations at different altitudes was, on average, 19.2, and across the 17 Czech units it was 25.8 alleles. The mean expected heterozygosity over these loci was much more similar in Tyrol populations (0.87) and in Czech units (0.86). A higher level of expected heterozygosity He = 0.934 was determined in Poland populations using three nuclear SSR markers [44]. Lower values were observed in Bosnia and Herzegovina populations (He ranged from 0.63 to 0.71).
Occurrences of null alleles were seen especially in loci PAAC19 and SpAGD1. A similar failure of amplifications for locus PAAC19 has been observed by Rungis et al. [24] and for locus SpAGD1 by Melnikova et al. [23]. Due to their recessive behavior, null alleles cause a shortage of heterozygote genotypes and result in incorrect estimates of allele frequencies. Experimental verification of null alleles is possible when new primers can be designed in microsatellite flanking regions, thereby resulting in a decrease of homozygotes [45]. With regard to the high number of different alleles (37 at locus PAAC19 and 38 at locus SpAGD1) across the studied Norway spruce units, we decided not to discard them from the evaluation; our main aim was to study the population diversities and, thus, it is better to use a greater number of polymorphic markers. Gömöry et al. [47] reported that null alleles could contribute to the overestimation of genetic differentiation among populations. We also calculated FST while excluding these two loci. In that case, we obtained slightly larger intervals of pairwise differentiation values (ranging from 0.005 to 0.031). The total level of genetic differentiation was 0.012 when these two loci were excluded. This compares to the previous result of 0.011 from nine loci. Among the geographically-closest subpopulations from the GZ 102–Orlické hory gene conservation unit, FST ranged in the lower values (0.005–0.012). Some results of other studies have shown that genetic differentiation could depend on the geographical distribution of sampling. Lower values of total FST estimation were found among three Austrian populations using SSR markers (FST = 0.002), thus indicating very small population differentiation. The spatial distance of these populations distributed at three different altitudes was only approximately 1000 m [25].
A higher total level of genetic differentiation (FST = 0.026) was observed among populations from Bosnia and Herzegovina that were spatially more distant [46]. In contrast to these results, however, in populations from the western Alps geographically located at shorter distances (only 40 km apart), meaningfully greater genetic differentiation was found (as high as FST = 0.089). The relatively high differentiation between geographically-close populations could be explained by their having originated from two distinct homogenous sources [48].
Low levels of genetic differentiation were observed among the Czech units, and the high genetic variation and heterozygosity within them is in conformity with similar results reported by other authors in previous studies for natural woody species populations, especially conifers. This could be explained by a mating system and high rate of migration provided by pollen and seed dispersal by wind or animals [6,48,49]. Another explanation may be associated with the changed geographic location of the populations due to extensive artificial forestation using Norway spruce since approximately the middle of the 19th century [50]. Genetic monitoring has been recognized on several international agreements and documents and can be an important tool for the protection of biodiversity. The use of genetic markers should be envisaged as a necessary complementary tool to demographic indicators for the complete assessment of a genetic resource. Genetic monitoring should concentrate on gene conservation units of such species, which should be advanced in a dynamic gene conservation scheme [51]. As the development of more powerful and affordable molecular markers and novel statistical and modelling tools is making genetic monitoring more feasible and cost-effective, it is reasonable to expect that an operational genetic monitoring system can be established for the dynamic conservation units in the near future [29].
5. Conclusions
The use of nuclear microsatellites has proven to be successful in detecting genetic variation in the studied units. In each naturally-regenerated population investigated, the allele number was high, thus indicating a higher level of genetic diversity that is comparable with that seen in Norway spruce populations of other European countries. Significant differences in allelic frequencies were found at single loci, but mean values from all loci showed low genetic differentiation between units. The shares of observed heterozygosity ranged from 0.65 to 0.80 and expected heterozygosity from 0.74 to 0.81. Significant knowledge for forestry management is that subpopulations from the gene conservation unit located in Orlické hory showed closer genetic similarity compared to that seen in other populations. Pairwise population FST values were greater than zero and so confirm the structuring of the investigated Czech populations. Structuring of populations was confirmed also by the subsequent finding in accordance with the results of Nei’s standard genetic distances, where the populations SM 01 and SM 05, both of Hurst ecotypes, were the closest to one another and the populations of mountain and alpine ecotypes were assembled into another group. The relation between the genetic differences observed in Norway spruce ecotypes and local adaptation needs to be further investigated. Acquiring new knowledge about the genetic structure of coniferous species populations, especially in relation to valuable ecotypes of Norway spruce, is very important in order to both maintain the ecological stability of forests and for the optimization of timber production. The developed procedures of genetic monitoring with DNA markers will be used in the amendment of forestry legislation and in state subsidy policy in the area of protection and reproduction of forest tree gene resources. These procedures for verifying the genetic quality of selected Czech gene conservation units will be used for subsequent monitoring strategies of other gene conservation units.
Supplementary Materials
The results from CERVUS 3.0.7, the results from GENEPOP 4.2, results from GenAlEx 6.501 and from STRUCTURE 2.3.4 are available online at http://www.mdpi.com/1999-4907/9/2/92/s1.
Acknowledgments
This work was supported by the projects of the Ministry of Agriculture of the Czech Republic—Resolution RO0117 (reference number 6779/2017-MZE-14151.) and no. NAZV QJ1530294.
Author Contributions
P.M. and H.C. conceived and designed the experiments, and discussed the results; P.M., H.C., and O.T. performed the experiments and analyzed the data; and H.C. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Musil, I.; Hamerník, J.; Leugnerová, G. Lesnická Dendrologie 1. Jehličnaté Dřeviny. [Forest Dendrology 1. Coniferous Trees]; The Czech Univerzity of Agriculture Prague: Praha, Czech Republic, 2003. [Google Scholar]
- Směrnice pro uznávání a zabezpečení zdrojů reprodukčního materiálu lesních dřevin a pro jeho přenos. [Directives for the Recognition and Security of Forest Resources and the Transfer of Forest Resources]; Ministry of Forestry and Water Management and Woodworking Industry of the Czechoslovak Republic: Praha, Czech Republic, 1988.
- Úradníček, L.; Maděra, P.; Tichá, S.; Koblížek, J. Dřeviny České Republiky [Woody Species of the Czech Republic]; Nakladatelství a vydavatelství Lesnická práce, s.r.o.: Kostelec nad Černými lesy, Czech Republic, 2009. [Google Scholar]
- Ministry of Agriculture of the Czech Republic. Information on Forests and Forestry in the Czech Republic by 2014. Available online: http://eagri.cz/public/web/file/433136/ZZ2014AJ_16112015.pdf (accessed on 27 June 2016).
- Šrámek, V.; Neudertová Hellebrandová, K. Mapy ohrožení smrkových porostů suchem jako nástroj identifikace rizikových oblastí [Maps of drought risk for Norway spruce stands as a decision tool indicating threatened regions in the Czech Republic: Short communication]. Rep. For. Res. 2016, 61, 305–309. [Google Scholar]
- Maghuly, F.; Pinsker, W.; Praznik, W.; Fluch, S. Genetic diversity in managed subpopulations of Norway spruce [Picea abies (L). Karst.]. For. Ecol. Manag. 2006, 222, 266–271. [Google Scholar]
- Hampe, A.; Petit, R.J. Conserving biodiversity under climate change: The rear edge matters. Ecol. Lett. 2005, 8, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Neale, D.B.; Kremer, A. Forest tree genomics: Growing resources and applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Whitham, T.G.; Bailey, J.K.; Schweitzer, J.A.; Shuster, S.M.; Bangert, R.K.; LeRoy, C.J.; Lonsdorf, E.V.; Allan, G.J.; DiFazio, S.P.; Potts, B.M.; et al. A framework for community and ecosystem genetics from genes to ecosystems. Nat. Rev. Genet. 2006, 7, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Geburek, T. Genetic variation of Norway spruce (Picea abies [L.] Karst.) populations in Austria. III. Macrospatial allozyme patterns of high elevation populations. For. Genet. 1999, 6, 201–211. [Google Scholar]
- Konnert, M. Genetic variation of Picea abies in southern Germany as determined using isozyme and STS markers. Dendrobiology 2009, 61, 131–136. [Google Scholar]
- Schubert, R.; Mueller-Starck, G.; Riegel, R. Development of EST-PCR markers and monitoring their intrapopulational genetic variation in Picea abies (L.) Karst. Theor. Appl. Genet. 2001, 103, 1223–1231. [Google Scholar]
- Bozhko, M.; Riegel, R.; Schubert, R.; Müller-Starck, G. A cyclophilin gene marker confirming geographical differentiation of Norway spruce populations and indicating viability response on excess soil-born salinity. Mol. Ecol. 2003, 12, 3147–3155. [Google Scholar] [CrossRef] [PubMed]
- Maghuly, F.; Burg, K.; Pinsker, W.; Nittinger, F.; Praznik, W.; Fluch, S. Short Note: Development of mitochondrial markers for population genetics of Norway Spruce [Picea abies (L). Karst]. Silvae Genet. 2008, 57, 41–44. [Google Scholar] [CrossRef]
- Tollefsrud, M.M.; Sønstebø, J.H.; Brochmann, C.; Johnsen, Ø.; Skroppa, T.; Vendramin, G.G. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European. Picea abies. Heredity 2009, 102, 549–562. [Google Scholar]
- Paglia, G.P.; Olivieri, A.M.; Morgante, M. Towards second-generation STS (sequence-tagged sites) linkage maps in conifers: A genetic map of Norway spruce (Picea abies K.). Mol. Gen. Genet. 1998, 258, 466–478. [Google Scholar]
- Perry, D.J.; Isabel, N.; Bousquet, J. Sequence-tagged-site (STS) markers of arbitrary genes: The amount and nature of variation revealed in Norway spruce. Heredity 1999, 83, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Acheré, V.; Favre, J.M.; Besnard, G.; Jeandroz, S. Genomic organization of molecular differentiation in Norway spruce (Picea abies). Mol. Ecol. 2005, 14, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Uebbing, S.; Gyllenstrand, N.; Lagercrantz, U.; Lascoux, M.; Källman, T. Sequencing of the needle transcriptome from Norway spruce (Picea abies Karst. L.) reveals lower substitution rates, but similar selective constraints in gymnosperms and angiosperms. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Romšáková, I.; Foffová, E.; Kmeť, J.; Longauer, R.; Pacalaj, M.; Gömöry, D. Nucleotide polymorphisms related to altitude and physiological traits in contrasting provenances of Norway spruce (Picea abies). Biologia 2012, 67, 909–916. [Google Scholar] [CrossRef]
- Pfeiffer, A.M.; Oliviery, A.M.; Morgante, M. Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 1997, 40, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Scotti, I.; Magni, F.; Fink, R.; Powell, W.; Binelli, G.; Hedley, P.E. Microsatellite repeats are not randomly distributed within Norway spruce (Picea abies K.) expressed sequences. Genome 2000, 43, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Melnikova, M.N.; Petrov, N.B.; Lomov, A.A.; la Porta, N.; Politov, D.V. Testing of Microsatellite Primers with Different Populations of Eurasian Spruces Picea abies (L.) Karst. and Picea obovata Ledeb. Rus. J. Genet. 2012, 48, 562–566. [Google Scholar] [CrossRef]
- Rungis, D.; Bérubé, Y.; Zhang, J.; Ralph, S.; Ritland, C.E.; Ellis, B.E.; Douglas, C.; Bohlmann, J.; Ritland, K. Robust simple sequence repeat markers for spruce (Picea spp.) from expressed sequence tags. Theor. Appl. Genet. 2004, 109, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Unger, G.M.; Konrad, H.; Geburek, T. Does spatial genetic structure increase with altitude? An answer from Picea abies in Tyrol, Austria. Plant Syst. Evol. 2011, 292, 133–141. [Google Scholar]
- Pastorelli, R.; Smulders, M.J.M.; VAN’T Westende, W.P.C.; Vosman, B.; Giannini, R.; Vettori, C.; Vendramin, G.G. Characterization of microsatellite markers in Fagus sylvatica L. and Fagus orientalis Lipsky. Mol. Ecol. 2003, 3, 76–78. [Google Scholar] [CrossRef]
- Scotti, I.; Paglia, G.; Magni, F.; Morgante, M. Population genetics (Picea abies Karst.) at regional scale: Sensitivity of different microsatellite motif classes in detecting differentiation. Ann. For. Sci. 2006, 63, 485–491. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterborn, A.; Zuccolo, A.; Lin, Y.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Koskela, J.; Lefèvre, F.; Schueler, S. Translating conservation genetics into management: Pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol. Conserv. 2013, 157, 39–49. [Google Scholar] [CrossRef]
- Van Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–392. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; von Holdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Harfouche, A.; Meilan, R.; Altman, A. Molecular and physiological responses to abiotic stress in forest trees and their relevance to tree improvement. Tree Physiol. 2014, 34, 1181–1198. [Google Scholar]
- Nowakowska, J.A. Mitochondrial and nuclear DNA differentiation of Picea abies populations in Poland. Dendrobiology 2009, 61, 119–129. [Google Scholar]
- Nascimento de Sousa, S.; Finkeldey, R.; Gailing, O. Experimental verification of microsatellite null alleles in Norway spruce (Picea abies [L.] Karst.): Implications for population genetic studies. Plant Mol. Biol. Rep. 2005, 23, 113–119. [Google Scholar] [CrossRef]
- Cvjetković, B.; Konnert, M.; Fussi, B.; Mataruga, M.; Šijačić-Nikolić, M.; Daničić, V.; Lučić, A. Norway spruce (Picea abies Karst.) variability in progeny tests in Bosnia and Herzegovina. Genetika 2017, 49, 259–272. [Google Scholar] [CrossRef]
- Gömöry, D.; Ditmarová, L.; Hrivnák, M.; Jamnická, G.; Kmet, J.; Krajmerová, D.; Kurjak, D. Differentiation in phenological and physiological traits in European beech (Fagus sylvatica L.). Eur. J. For. Res. 2015, 134, 1075–1085. [Google Scholar] [CrossRef]
- Meloni, M.; Perini, D.; Binelli, G. The distribution of genetic variation in Norway spruce (Picea abies [L.] Karst.) populations in the western Alps. J. Biogeogr. 2007, 34, 929–938. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Svoboda, P. Lesní dřeviny a jejich porosty. Část I. [Forest Tree Species and Their Stands. Part I]; Státní zemědělské nakladatelství: Prague, Czech Republic, 1953. [Google Scholar]
- Aravanopoulos, F.A. Genetic monitoring in natural perennial plant populations. Botany 2011, 89, 75–81. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).