Transcriptome Characterization of the Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Experiment
2.2. RNA Extraction, Library Preparation, and Transcriptome Sequencing
2.3. Data Processing, Assembly, and Annotation
2.4. qRT-PCR Analysis for Gene Expression
3. Results and Discussion
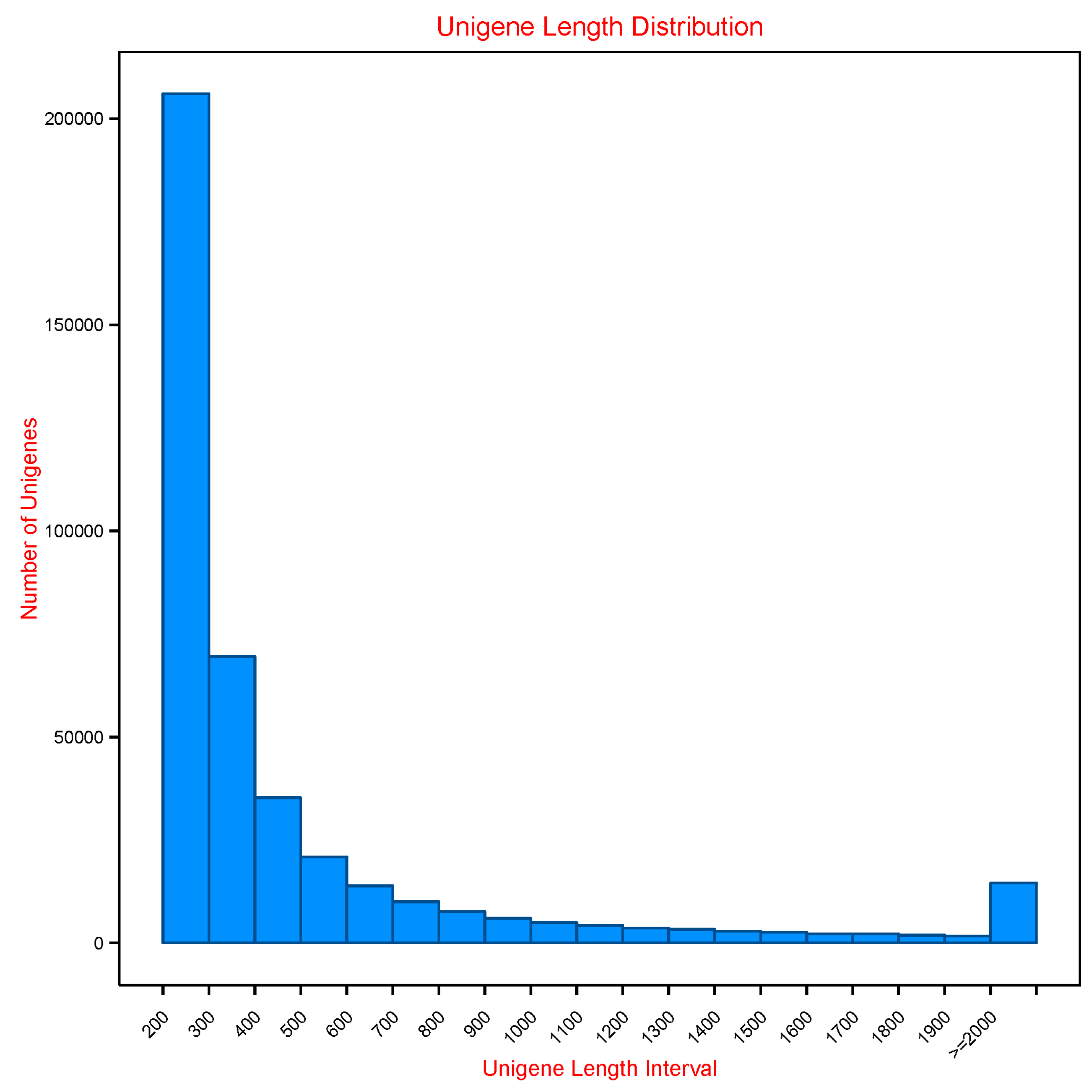
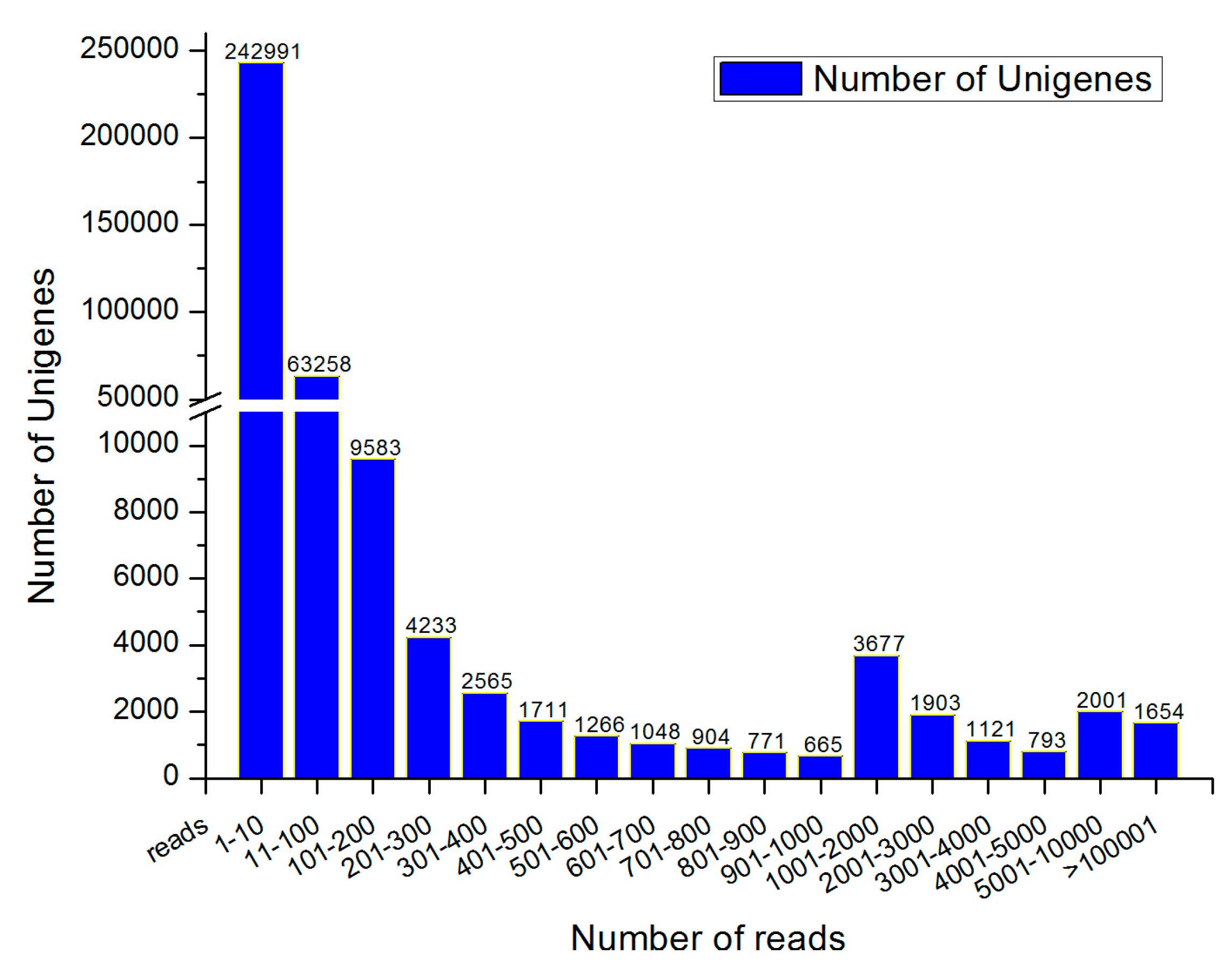
3.1. Sequencing and De Novo Assembly
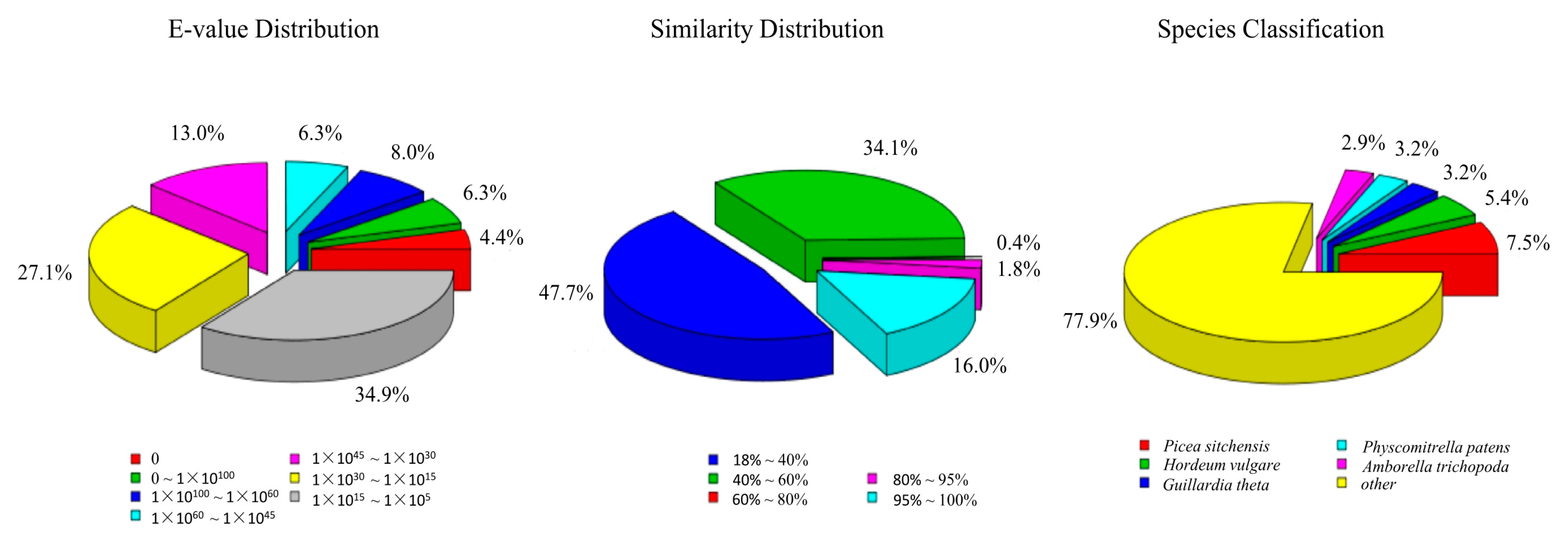
3.2. Annotation of Predicted Proteins
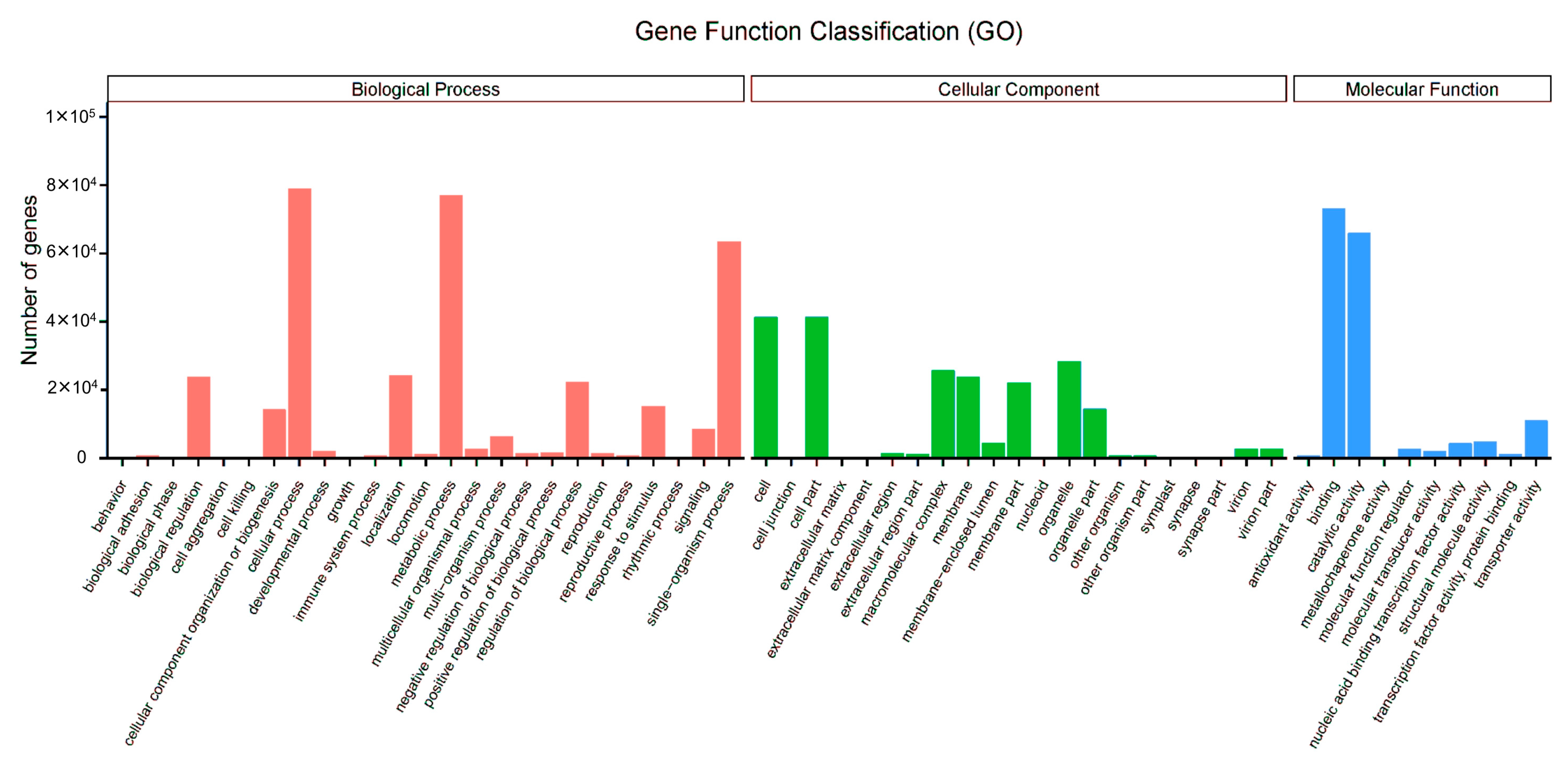
3.3. GO Assignments
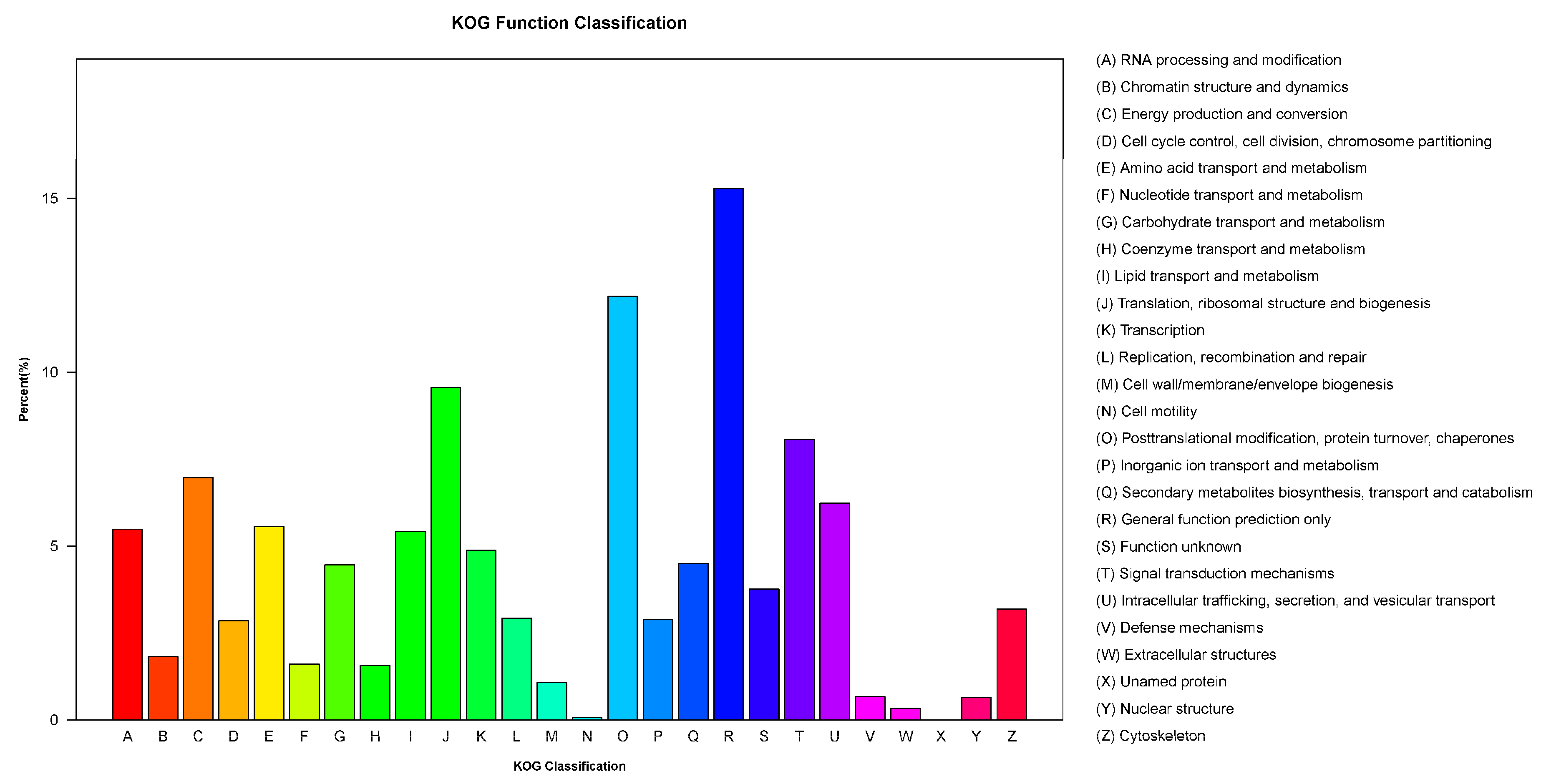
3.4. KOG Classification
3.5. KEGG Pathway Analysis
3.6. Candidate Genes Involved in Inorganic Phosphate Transport
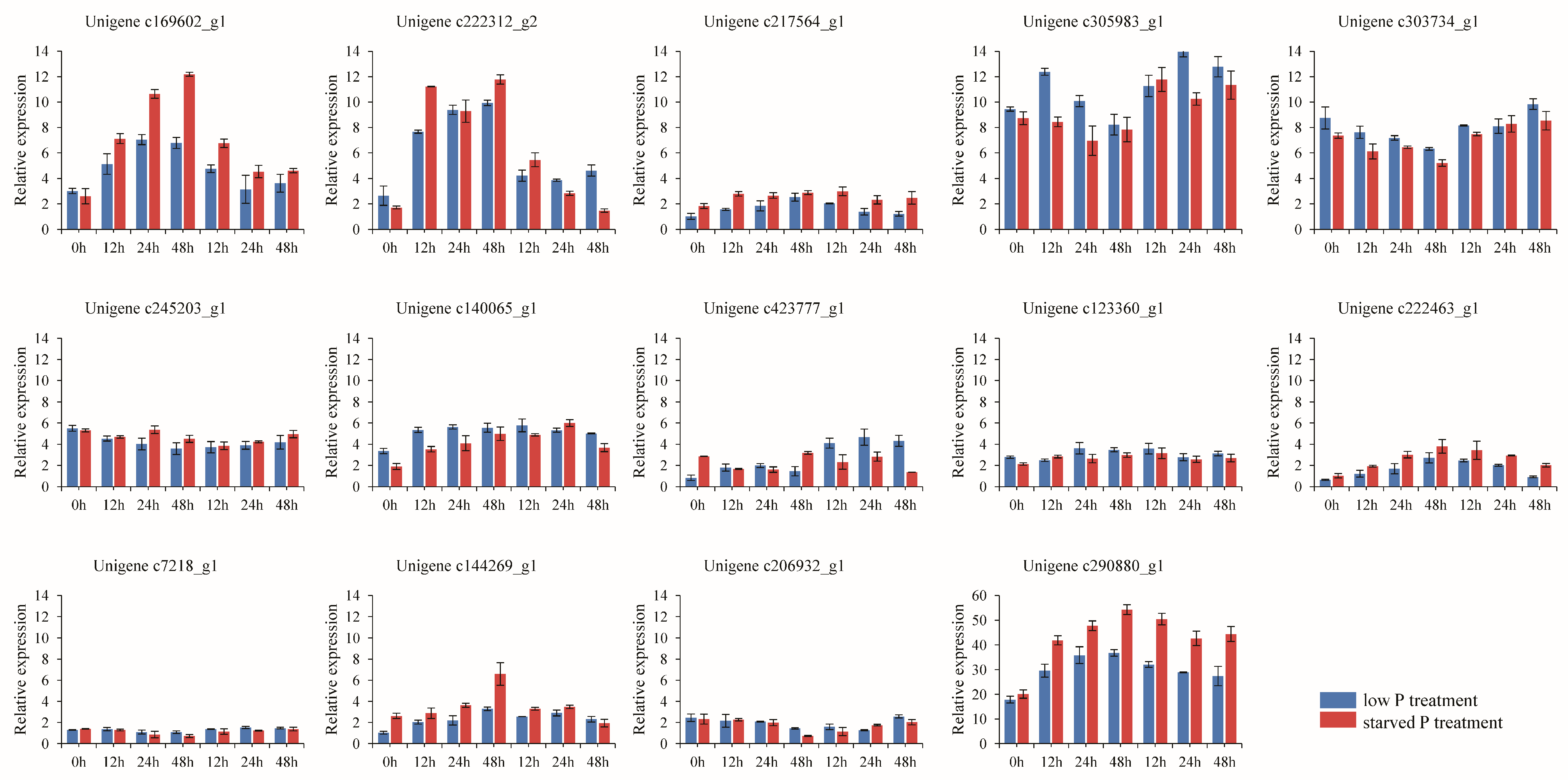
3.7. Gene Expression Analysis by qRT-PCR
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, X.Q.; Heal, K.V.; Liu, A.Q.; Jarvis, P.G. Nutrient cycling and distribution in different-aged plantations of chinese fir in southern china. For. Ecol. Manag. 2007, 243, 61–74. [Google Scholar] [CrossRef]
- Bian, L.M.; Shi, J.S.; Zheng, R.H.; Chen, J.H.; Wu, H.X. Genetic parameters and genotype-environment interactions of chinese fir (Cunninghamia lanceolata) in fujian province. Can. J. For. Res. 2014, 44, 582–592. [Google Scholar] [CrossRef]
- Yang, Y.S.; Guo, J.F.; Chen, G.S.; Xie, J.S.; Gao, R.; Li, Z.; Jin, Z. Carbon and nitrogen pools in chinese fir and evergreen broadleaved forests and changes associated with felling and burning in mid-subtropical China. For. Ecol. Manag. 2005, 216, 216–226. [Google Scholar] [CrossRef]
- Fan, H.B.; Hong, W. Estimation of dry deposition and canopy exchange in chinese fir plantations. For. Ecol. Manag. 2001, 147, 99–107. [Google Scholar] [CrossRef]
- Tian, D.L.; Xiang, W.H.; Chen, X.Y.; Yan, W.D.; Fang, X.; Kang, W.X.; Dan, X.W.; Peng, C.H.; Peng, Y.Y. A long-term evaluation of biomass production in first and second rotations of chinese fir plantations at the same site. Forestry 2011, 84, 411–418. [Google Scholar] [CrossRef]
- Zhang, X.-Q.; Kirschbaum, M.U.F.; Hou, Z.H.; Guo, Z.H. Carbon stock changes in successive rotations of chinese fir (Cunninghamia lanceolata (lamb) hook) plantations. For. Ecol. Manag. 2004, 202, 131–147. [Google Scholar] [CrossRef]
- Xu, X.J.; Timmer, V.R. Growth and nitrogen nutrition of chinese fir seedlings exposed to nutrient loading and fertilization. Plant Soil 1999, 216, 83–91. [Google Scholar] [CrossRef]
- Wu, P.F.; Tigabu, M.; Ma, X.Q.; Oden, P.C.; He, Y.L.; Yu, X.T.; He, Z.Y. Variations in biomass, nutrient contents and nutrient use efficiency among chinese fir provenances. Silvae Genet. 2011, 60, 95–105. [Google Scholar] [CrossRef]
- Chen, H.J. Phosphatase activity and p fractions in soils of an 18-year-old chinese fir (Cunninghamia lanceolata) plantation. For. Ecol. Manag. 2003, 178, 301–310. [Google Scholar] [CrossRef]
- Wu, P.F.; Ma, X.Q.; Tigabu, M.; Wang, C.; Liu, A.Q.; Oden, P.C. Root morphological plasticity and biomass production of two chinese fir clones with high phosphorus efficiency under low phosphorus stress. Can. J. For. Res. 2011, 41, 228–234. [Google Scholar] [CrossRef]
- Shenoy, V.; Kalagudi, G. Enhancing plant phosphorus use efficiency for sustainable cropping. Biotechnol. Adv. 2005, 23, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.S.; Niklas, K.J.; Liu, Y.; Fang, X.M.; Wan, S.Z.; Wang, H.M. Nitrogen and phosphorus additions alter nutrient dynamics but not resorption efficiencies of chinese fir leaves and twigs differing in age. Tree Physiol. 2015, 35, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.H.; Wu, P.F.; Chen, N.L.; Wang, P.; Ma, X.Q. Chinese fir root response to spatial and temporal heterogeneity of phosphorus availability in the soil. Can. J. For. Res. 2015, 45, 402–410. [Google Scholar] [CrossRef]
- Chen, Y.X.; He, Y.F.; Kumar, S.; Fu, Q.L.; Tian, G.M.; Lin, Q. Soil phosphorus status under restored plant covers established to control land degradation in the red soil region of south china. J. Soil Water Conserv. 2002, 57, 381–387. [Google Scholar]
- Tang, Y.C.; Zhang, X.Y.; Li, D.D.; Wang, H.M.; Chen, F.S.; Fu, X.L.; Fang, X.M.; Sun, X.M.; Yu, G.R. Impacts of nitrogen and phosphorus additions on the abundance and community structure of ammonia oxidizers and denitrifying bacteria in chinese fir plantations. Soil Biol. Biochem. 2016, 103, 284–293. [Google Scholar] [CrossRef]
- Wang, F.E.; Chen, Y.X.; Tian, G.M.; Kumar, S.; He, Y.F.; Fu, Q.L.; Lin, Q. Microbial biomass carbon, nitrogen and phosphorus in the soil profiles of different vegetation covers established for soil rehabilitation in a red soil region of southeastern china. Nutr. Cycl. Agroecosyst. 2004, 68, 181–189. [Google Scholar] [CrossRef]
- Hammond, J.P.; White, P.J. Sucrose transport in the phloem: Integrating root responses to phosphorus starvation. J. Exp. Bot. 2008, 59, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.-N.; Zhu, Y.-G.; Liu, W.-J.; Smith, S.E. Arsenate uptake and translocation in seedlings of two genotypes of rice is affected by external phosphate concentrations. Aquat. Bot. 2005, 83, 321–331. [Google Scholar] [CrossRef]
- Loth-Pereda, V.; Orsini, E.; Courty, P.-E.; Lota, F.; Kohler, A.; Diss, L.; Blaudez, D.; Chalot, M.; Nehls, U.; Bucher, M. Structure and expression profile of the phosphate pht1 transporter gene family in mycorrhizal Populus trichocarpa. Plant Physiol. 2011, 156, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- Kavka, M.; Polle, A. Phosphate uptake kinetics and tissue-specific transporter expression profiles in poplar (Populus × canescens) at different phosphorus availabilities. BMC Plant Biol. 2016, 16, 206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Meng, S.; Li, M.; Zhao, Z. Genomic identification and expression analysis of the phosphate transporter gene family in poplar. Front. Plant Sci. 2016, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Nussaume, L.; Kanno, S.; Javot, H.; Marin, E.; Nakanishi, T.M.; Thibaud, M.-C. Phosphate import in plants: Focus on the pht1 transporters. Front. Plant Sci. 2011, 2, 83. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-H.; Xu, L.-L.; Tong, Z.-K.; Lin, E.-P.; Liu, Q.-P.; Cheng, L.-J.; Zhu, M.-Y. De novo characterization of the chinese fir (Cunninghamia lanceolata) transcriptome and analysis of candidate genes involved in cellulose and lignin biosynthesis. BMC Genom. 2012, 13, 648. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Gao, Y.; Yang, L.; Shi, J. Identification and analysis of differentially expressed genes in differentiating xylem of chinese fir (Cunninghamia lanceolata) by suppression subtractive hybridization. Genome 2007, 50, 1141–1155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Liu, W.; Luo, Z.; Wang, P.; Zhang, Y.; Zheng, R.; Shi, J. Transcriptome characteristics and six alternative expressed genes positively correlated with the phase transition of annual cambial activities in chinese fir (Cunninghamia lanceolata (Lamb.) Hook). PLoS ONE 2013, 8, e71562. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Deng, Z.; Qin, B.; Liu, X.; Men, Z. De novo assembly and characterization of bark transcriptome using illumina sequencing and development of est-ssr markers in rubber tree (Hevea brasiliensis Muell. Arg.). BMC Genom. 2012, 13, 192. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from rna-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The kegg resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the blast2go suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for rna-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the kegg orthology (ko) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. Ngs qc toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Wan, L.; Chen, T.; Wan, Y.; He, X.; Lu, S.; Wang, Y.; Lin, J. The regulation of cambial activity in chinese fir (Cunninghamia lanceolata) involves extensive transcriptome remodeling. New Phytol. 2013, 199, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-Y.; Yang, H.; Wei, C.-L.; Yu, O.; Zhang, Z.-Z.; Jiang, C.-J.; Sun, J.; Li, Y.-Y.; Chen, Q.; Xia, T. Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom. 2011, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-M.; Zhao, L.; Larson-Rabin, Z.; Li, D.-Z.; Guo, Z.-H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef] [PubMed]
- Parchman, T.L.; Geist, K.S.; Grahnen, J.A.; Benkman, C.W.; Buerkle, C.A. Transcriptome sequencing in an ecologically important tree species: Assembly, annotation, and marker discovery. BMC Genom. 2010, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-Y.; Zhao, N.; Yang, B. Global transcriptome profiling of the pine shoot beetle, tomicus yunnanensis (coleoptera: Scolytinae). PLoS ONE 2012, 7, e32291. [Google Scholar] [CrossRef] [PubMed]
- Rigault, P.; Boyle, B.; Lepage, P.; Cooke, J.E.; Bousquet, J.; MacKay, J.J. A white spruce gene catalog for conifer genome analyses. Plant Physiol. 2011, 157, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A. The norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The gene ontology project in 2008. Nucleic Acids Res. 2008, 36, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Mossio, M.; Montévil, M.; Longo, G. Theoretical principles for biology: Organization. Prog. Biophys. Mol. Biol. 2016, 122, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The cog database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The cog database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Cancino-Rodezno, A.; Lozano, L.; Oppert, C.; Castro, J.I.; Lanz-Mendoza, H.; Encarnación, S.; Evans, A.E.; Gill, S.S.; Soberón, M.; Jurat-Fuentes, J.L. Comparative proteomic analysis of Aedes aegypti larval midgut after intoxication with cry11aa toxin from bacillus thuringiensis. PLoS ONE 2012, 7, e37034. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.; Grabherr, M.G.; Guttman, M.; Trapnell, C. Computational methods for transcriptome annotation and quantification using rna-seq. Nat. Methods 2011, 8, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Baseler, W.A.; Thapa, D.; Jagannathan, R.; Dabkowski, E.R.; Croston, T.L.; Hollander, J.M. Mir-141 as a regulator of the mitochondrial phosphate carrier (slc25a3) in the type 1 diabetic heart. Am. J. Physiol. Cell Physiol. 2012, 303, C1244–C1251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Miao, Q.; Sun, D.; Yang, G.; Wu, C.; Huang, J.; Zheng, C. The mitochondrial phosphate transporters modulate plant responses to salt stress via affecting atp and gibberellin metabolism in arabidopsis thaliana. PLoS ONE 2012, 7, e43530. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wan, X.; Zhu, W.; Sun, D.; Zheng, C.; Liu, P.; Huang, J. Overexpression of mitochondrial phosphate transporter 3 severely hampers plant development through regulating mitochondrial function in arabidopsis. PLoS ONE 2015, 10, e0129717. [Google Scholar] [CrossRef] [PubMed]
- Trentmann, O.; Jung, B.; Neuhaus, H.E.; Haferkamp, I. Nonmitochondrial atp/adp transporters accept phosphate as third substrate. J. Biol. Chem. 2008, 283, 36486–36493. [Google Scholar] [CrossRef] [PubMed]
- Perrin, J.; Mortier, M.; Jacomin, A.-C.; Viargues, P.; Thevenon, D.; Fauvarque, M.-O. The nonaspanins tm9sf2 and tm9sf4 regulate the plasma membrane localization and signalling activity of the peptidoglycan recognition protein pgrp-lc in drosophila. J. Innate Immun. 2014, 7, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Ceasar, S.A.; Palmer, A.J.; Paterson, J.B.; Qi, W.; Muench, S.P.; Baldwin, S.A. Replace, reuse, recycle: Improving the sustainable use of phosphorus by plants. J. Exp. Bot. 2015, 66, 3523–3540. [Google Scholar] [CrossRef] [PubMed]
- López-Arredondo, D.L.; Leyva-González, M.A.; González-Morales, S.I.; López-Bucio, J.; Herrera-Estrella, L. Phosphate nutrition: Improving low-phosphate tolerance in crops. Annu. Rev. Plant Biol. 2014, 65, 95–123. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Jung, J.-Y. Phosphate transporters. Annu. Plant Rev. Phosphorus Metab. Plants 2015, 48, 125. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 1 (%) | Q30 2 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| Root | 80,914,328 | 76,829,210 | 11.52 G | 0.02 | 96.64 | 92.18 | 47.94 |
| Stem | 86,439,146 | 82,165,990 | 12.32 G | 0.01 | 96.7 | 92.31 | 44.22 |
| Leaves | 81,365,604 | 77,534,078 | 11.63 G | 0.01 | 96.79 | 92.45 | 44.22 |
| Summary | 248,719,078 | 236,529,278 | 35.47 G | 45.46 |
| Fragment Length (bp) | Transcripts | Unigenes |
|---|---|---|
| 200–500 bp | 338,030 | 311,104 |
| 500–1000 bp | 74,144 | 58,295 |
| 1000–2000 bp | 43,311 | 29,834 |
| >2000 bp | 23,550 | 14,573 |
| Total | 479,035 | 413,806 |
| Minimal length (bp) | 201 | 201 |
| Maximal length (bp) | 17,835 | 17,835 |
| N50 (bp) | 891 | 671 |
| Mean length (bp) | 591 | 520 |
| Total Nucleotides | 282,892,631 | 215,269,640 |
| Gene_ID | Gene Length (bp) | FPKM Leaf | FPKM Root | FPKM Stem | Function Annotation |
|---|---|---|---|---|---|
| c169602_g1 | 1102 | 0 | 1.73 | 0 | MFS transporter, PHO48, high affinity inorganic phosphate transporter |
| c222312_g2 | 451 | 0 | 1.16 | 0 | MFS transporter, PHO48, high affinity inorganic phosphate transporter |
| c217564_g1 | 2767 | 1.86 | 0.28 | 0.54 | MFS transporter, ACS family, SLC17A, inorganic diphosphate transmembrane transporter activity |
| c305983_g1 | 247 | 0 | 6.86 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c290880_g1 | 263 | 0 | 6.23 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c303734_g1 | 272 | 0 | 4.89 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c245203_g1 | 271 | 0 | 3.35 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c140065_g1 | 422 | 0 | 1.86 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c423777_g1 | 466 | 0 | 1.26 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c123360_g1 | 990 | 0 | 1.02 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c222463_g1 | 2841 | 4.57 | 0.14 | 2.49 | Inorganic phosphate transporter 2-1 (Medicago truncatula) |
| c7218_g1 | 478 | 0 | 0.51 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c144269_g1 | 532 | 0 | 0.94 | 0 | SLC20A PIT, inorganic phosphate transmembrane transporter activity |
| c206932_g1 | 3127 | 0.8 | 0.61 | 0.95 | Inorganic phosphate transporter 2-1 (Medicago truncatula) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Su, S.-S.; Wu, P.-F.; Cameron, K.M.; Zhang, Y.; Chen, W.-T.; Ma, X.-Q. Transcriptome Characterization of the Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes. Forests 2017, 8, 420. https://doi.org/10.3390/f8110420
Li M, Su S-S, Wu P-F, Cameron KM, Zhang Y, Chen W-T, Ma X-Q. Transcriptome Characterization of the Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes. Forests. 2017; 8(11):420. https://doi.org/10.3390/f8110420
Chicago/Turabian StyleLi, Ming, Suo-Suo Su, Peng-Fei Wu, Kenneth M. Cameron, Ying Zhang, Wan-Ting Chen, and Xiang-Qing Ma. 2017. "Transcriptome Characterization of the Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes" Forests 8, no. 11: 420. https://doi.org/10.3390/f8110420
APA StyleLi, M., Su, S.-S., Wu, P.-F., Cameron, K. M., Zhang, Y., Chen, W.-T., & Ma, X.-Q. (2017). Transcriptome Characterization of the Chinese Fir (Cunninghamia lanceolata (Lamb.) Hook.) and Expression Analysis of Candidate Phosphate Transporter Genes. Forests, 8(11), 420. https://doi.org/10.3390/f8110420