
Genome-Wide Resequencing Revealed the Genetic Diversity of Fraxinus platypoda Oliv. in Northwestern China


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Sequencing
2.2. SNP Calling
2.3. Population Genetic Structure Analysis
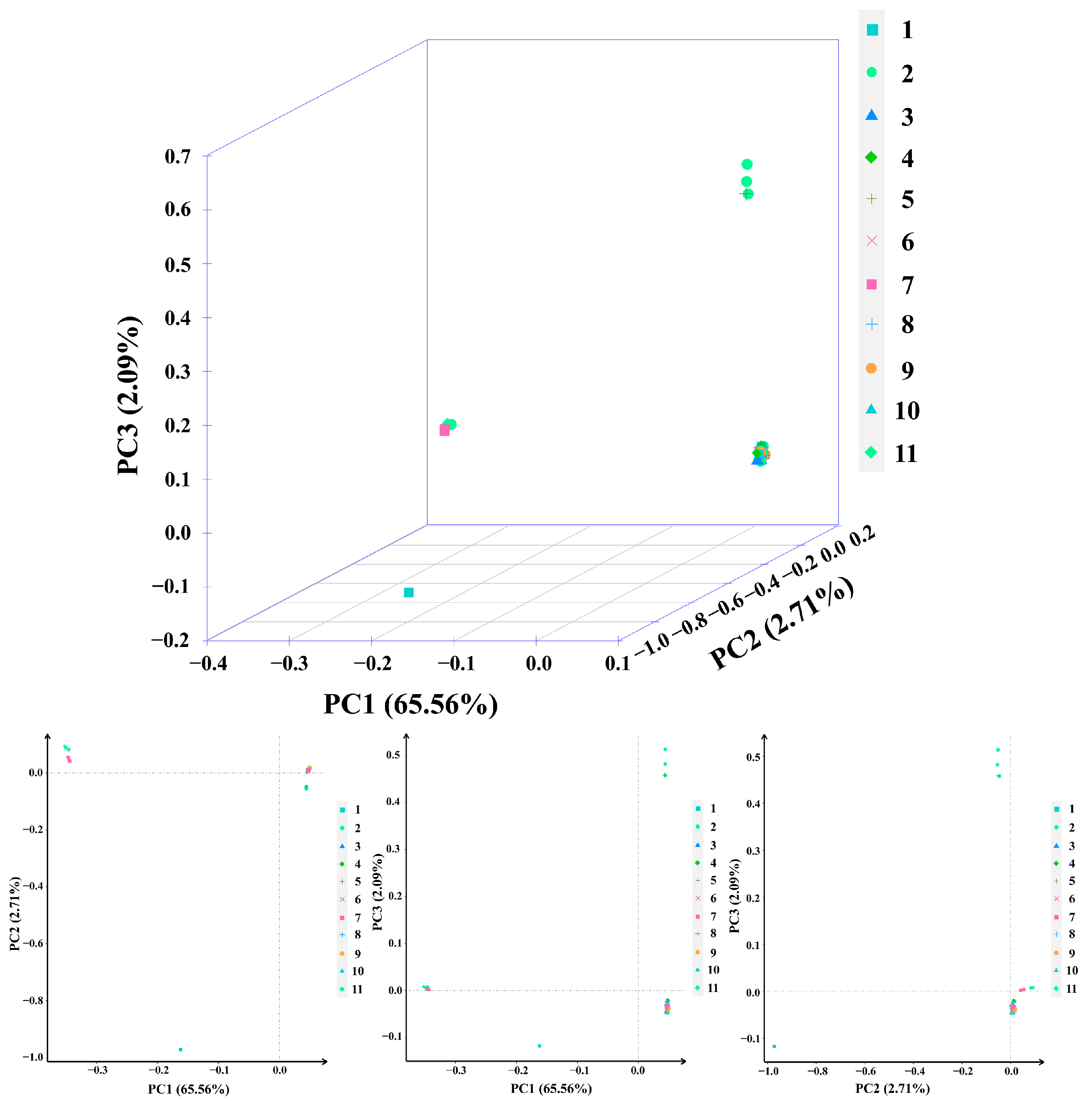
2.4. Principal Component Analysis
2.5. Linkage Disequilibrium Analysis
2.6. The Analysis of Molecular Variance and Genetic Diversity
2.7. Isolation by Distance (IBD)
3. Results
3.1. Quality Control and the Filtration of Data
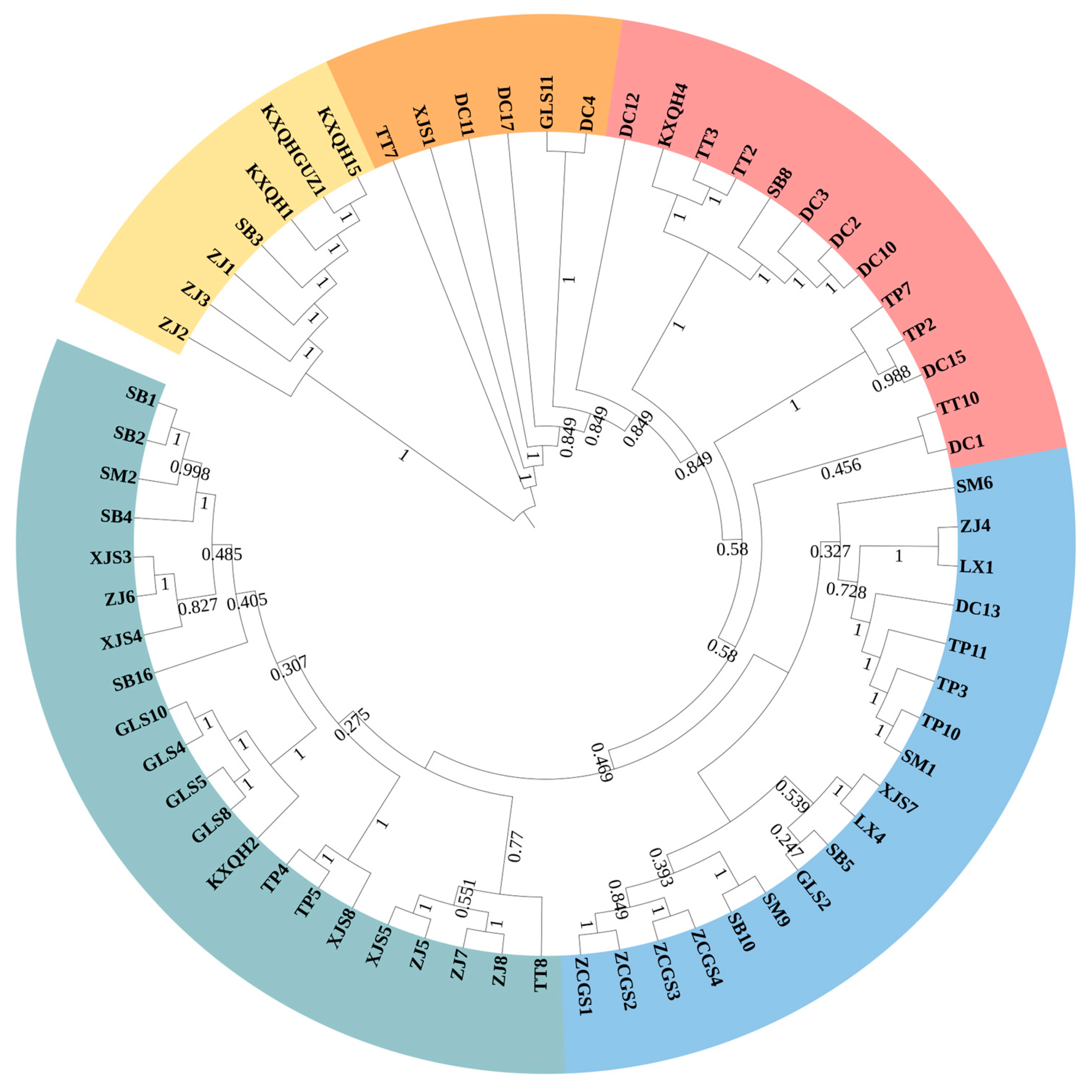
3.2. Phylogenetic Relationship
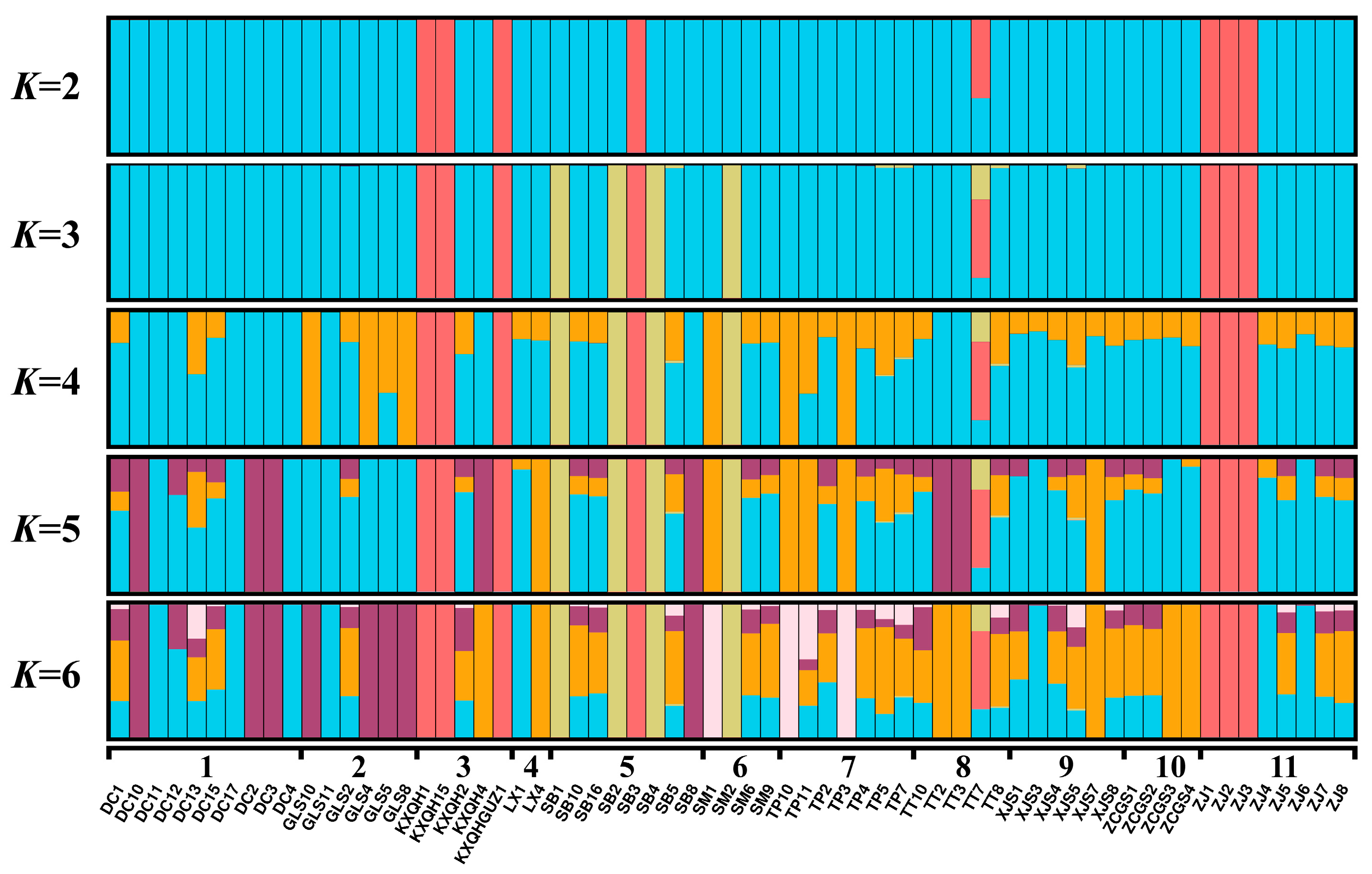
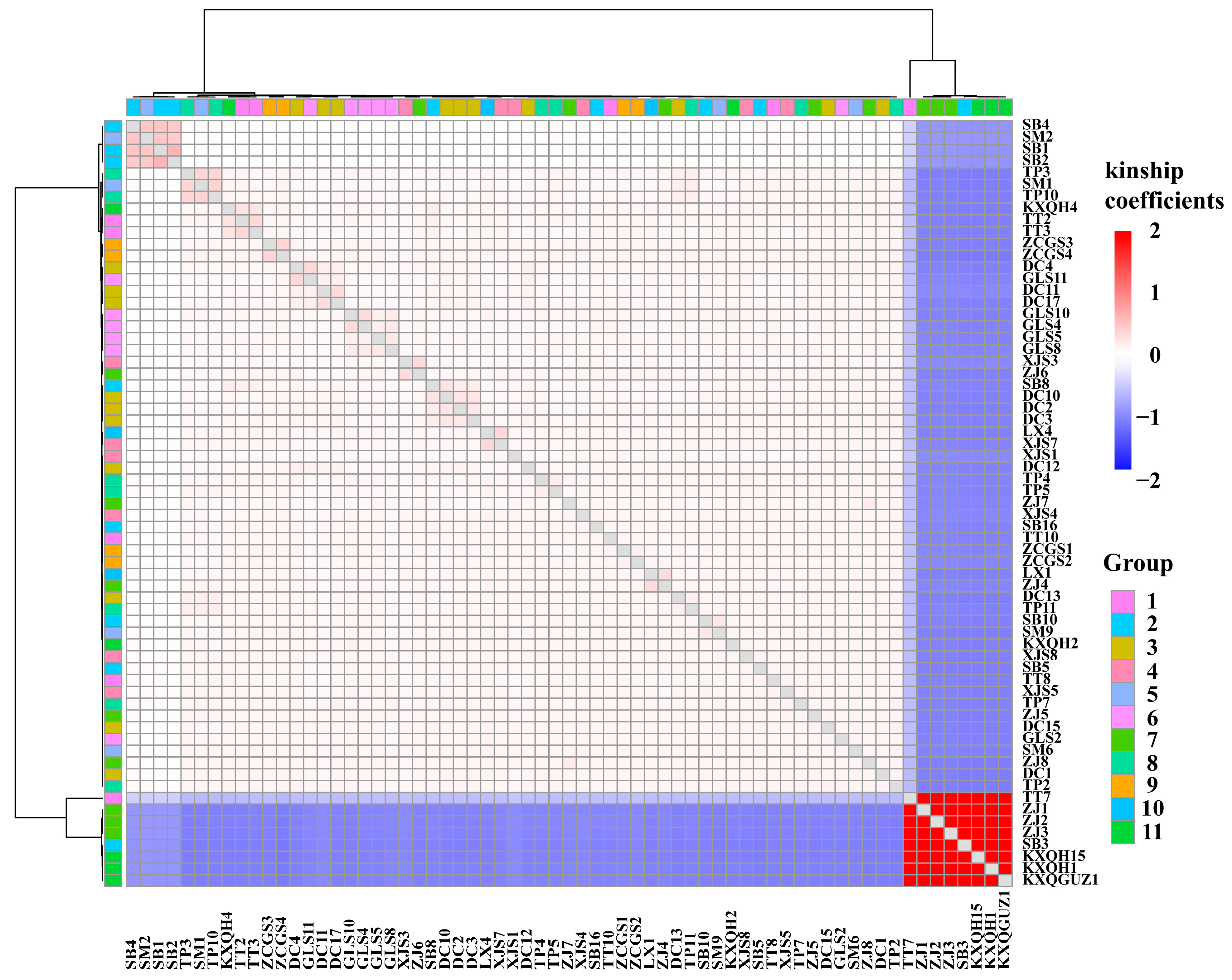
3.3. Population Genetic Structure
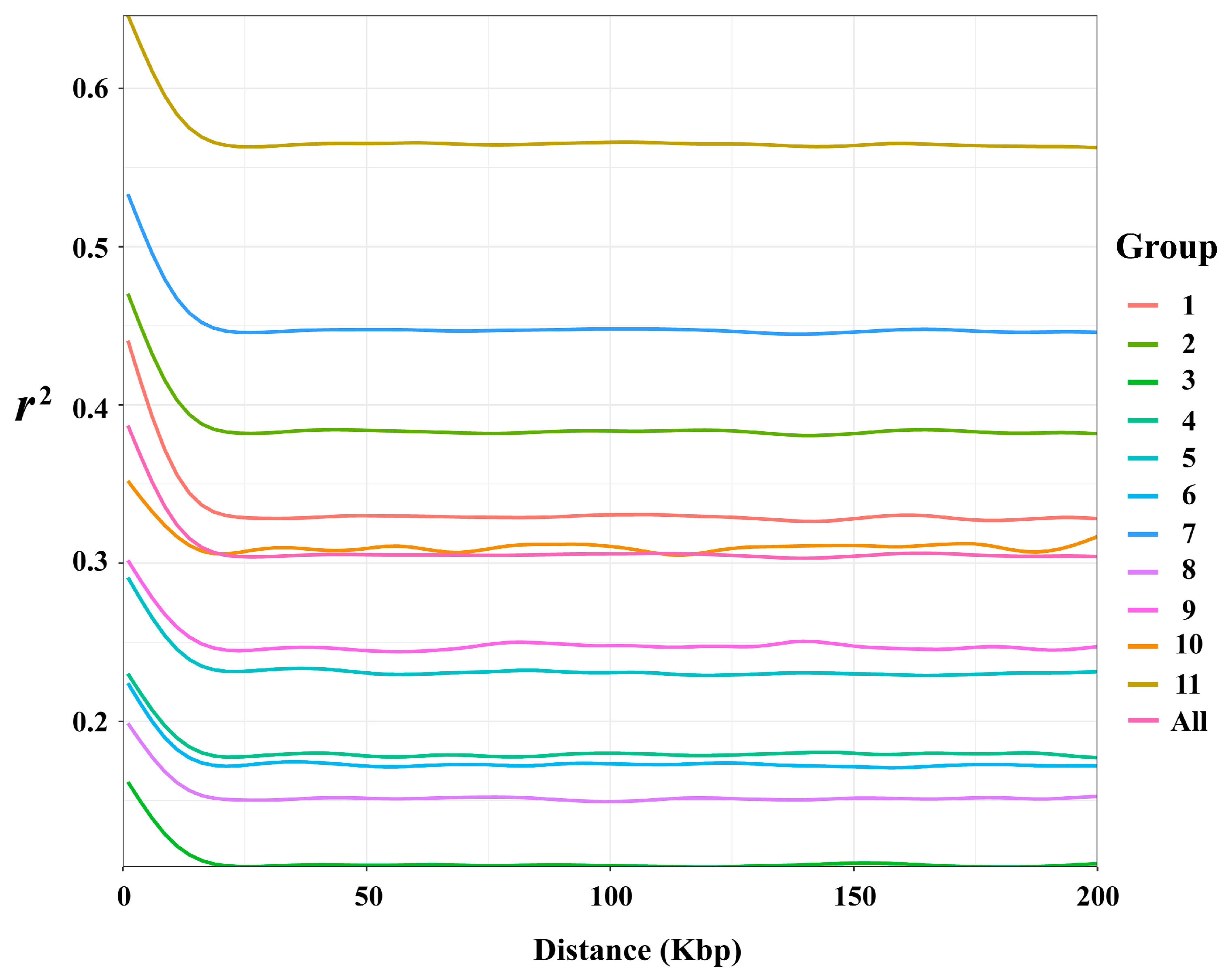
3.4. Linkage Disequilibrium Decay
3.5. Population Diversity Analysis
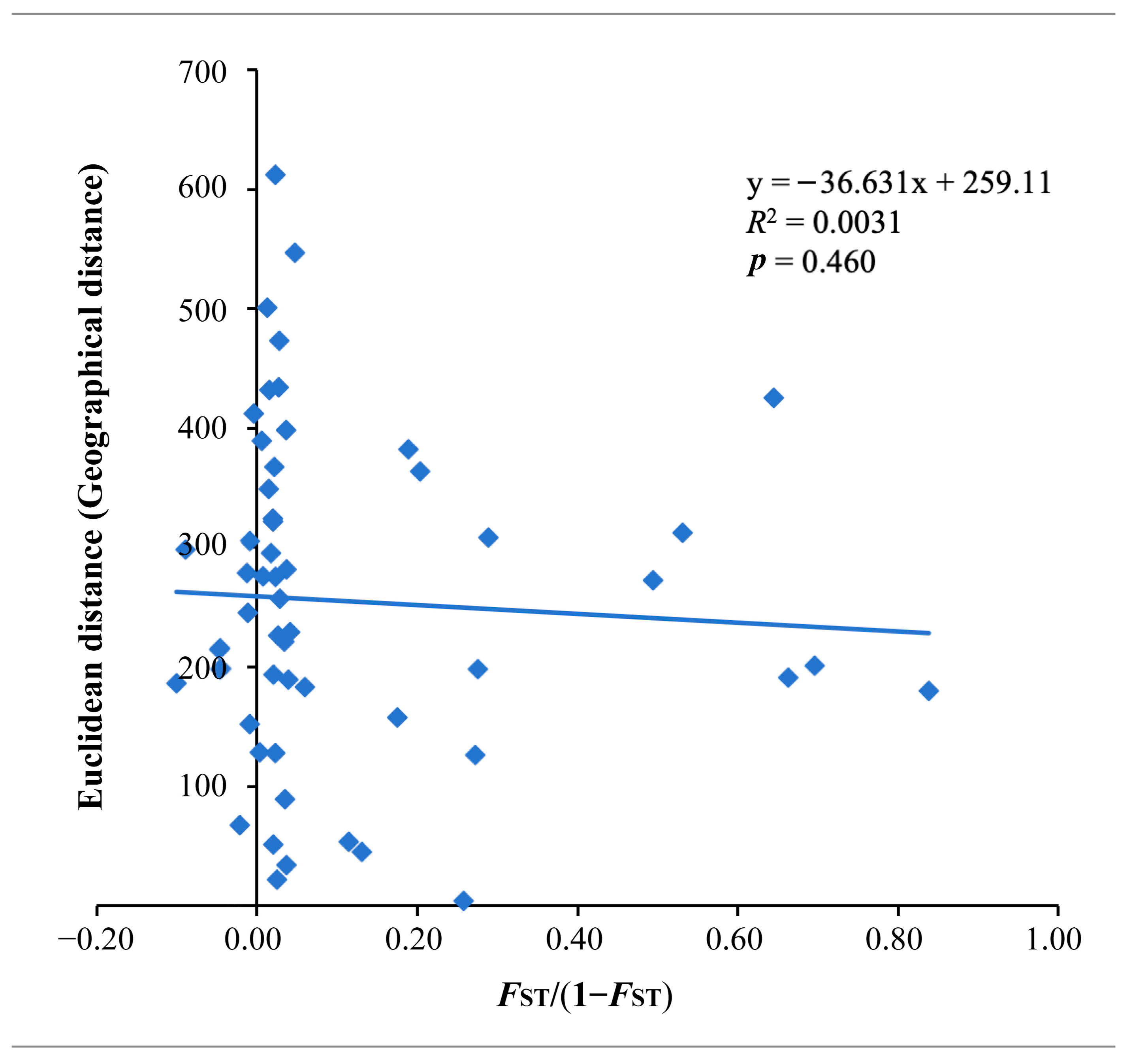
3.6. Isolation by Distance Analysis
4. Discussion
4.1. The Genetic Structure of F. platypoda
4.2. The Genetic Diversity of F. platypoda
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| F. platypoda | Fraxinus platypoda |
| NW | Northwestern |
| SNP | single nucleotide polymorphism |
| PCA | principal component analysis |
| He | expected heterozygosity |
| Ho | observed heterozygosity |
| MAF | minor allele frequency |
| PIC | polymorphism information content |
| GVM | Genome Variation Map |
References
- Rands, M.R.; Adams, W.M.; Bennun, L.; Butchart, S.M.; Clements, A.; Coomes, D.; Entwistle, A.; Hodge, I.; Kapos, V.; Scharlemann, J.W.; et al. Biodiversity conservation: Challenges beyond 2010. Science 2010, 329, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, S.L.; Fuller, R.A.; Brooks, T.M.; Watson, J.M. Biodiversity: The ravages of guns, nets and bulldozers. Nature 2016, 536, 143–145. [Google Scholar] [CrossRef]
- Haddad, N.M.; Brudvig, L.A.; Clobert, J.; Davies, K.F.; Gonzalez, A.; Holt, R.D.; Lovejoy, T.E.; Sexton, J.O.; Austin, M.P.; Collins, C.D.; et al. Habitat fragmentation and its lasting impact on Earth’s ecosystems. Sci. Adv. 2015, 1, e1500052. [Google Scholar] [CrossRef]
- Newbold, T.; Hudson, L.N.; Hill, S.L.; Contu, S.; Lysenko, I.; Senior, R.A.; Börger, L.; Bennett, D.J.; Choimes, A.; Collen, B.; et al. Global effects of land use on local terrestrial biodiversity. Nature 2015, 520, 45–50. [Google Scholar] [CrossRef]
- Lande, R. Genetics and demography in biological conservation. Science 1988, 241, 1455–1460. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Sork, V.L.; Smouse, P.E. Genetic Analysis of Landscape Connectivity in Tree Populations. Landsc. Ecol. 2006, 21, 821–836. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Yadav, L.K.; Bellis, D.; Smith, Z.C.; Ony, M.; Hale, C.; Richards, C.; Klingeman, W.E.; Staton, M.E.; Granger, J.J.; Hadziabdic, D. Genetic diversity and population structure of a rare flowering tree endemic to Appalachia, Stewartia ovata. Ecol. Evol. 2024, 14, e11547. [Google Scholar] [CrossRef]
- Young, A.; Boyle, T.; Brown, T. The population genetic consequences of habitat fragmentation for plants. Trends Ecol. Evol. 1996, 11, 413–418. [Google Scholar] [CrossRef]
- Yao, X.; Ye, Q.; Kang, M.; Huang, H. Microsatellite analysis reveals interpopulation differentiation and gene flow in the endangered tree Changiostyrax dolichocarpa (Styracaceae) with fragmented distribution in central China. New Phytol. 2007, 176, 472–480. [Google Scholar] [CrossRef]
- Rocchetti, G.A. Vascular Plant Extinction in Macaronesia: Biogeographical and Biological Drivers of Loss. Glob. Change Biol. 2025, 31, e70128. [Google Scholar] [CrossRef] [PubMed]
- Bacles, C.F.E.; Burczyk, J.; Lowe, A.J.; Ennos, R.A. Historical and contemporary mating patterns in remnant populations of the forest tree Fraxinus excelsior L. Evolution 2005, 59, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Semizer-Cuming, D.; Kjær, E.D.; Finkeldey, R. Gene flow of common ash (Fraxinus excelsior L.) in a fragmented landscape. PLoS ONE 2017, 12, e0186757. [Google Scholar] [CrossRef] [PubMed]
- Hinsinger, D.D.; Basak, J.; Gaudeul, M.; Cruaud, C.; Bertolino, P.; Frascaria-Lacoste, N.; Bousquet, J. The phylogeny and biogeographic history of ashes (Fraxinus, Oleaceae) highlight the roles of migration and vicariance in the diversification of temperate trees. PLoS ONE 2013, 8, e80431. [Google Scholar] [CrossRef]
- Li, E.; Wang, Y.; Liu, K.; Liu, Y.; Xu, C.; Dong, W.; Zhang, Z. Historical climate change and vicariance events contributed to the intercontinental disjunct distribution pattern of ash species (Fraxinus, Oleaceae). Commun. Biol. 2024, 7, 603. [Google Scholar] [CrossRef]
- Wallander, E. Systematics of Fraxinus (Oleaceae) and evolution of dioecy. Plant Syst. Evol. 2008, 273, 25–49. [Google Scholar] [CrossRef]
- Sakio, H. (Ed.) Fraxinus platypoda. In Long-Term Ecosystem Changes in Riparian Forest; Springer: Singapore, 2020; pp. 23–37. [Google Scholar]
- Zhao, X.X.; Ju, T.Z.; Wang, S.; Lu, X.; Wang, J.; Xie, S.; Zhang, S. Analysis of population structure characteristics and spatial distribution pattern of Fraxinus platypoda Oliv. in Xiaolongshan National Nature Reserve. Jiangsu Agric. Sci. 2018, 46, 235–239. [Google Scholar] [CrossRef]
- Cui, J.; Yang, Y.; Luo, S.; Wang, L.; Huang, R.; Wen, Q.; Han, X.; Miao, N.; Cheng, J.; Liu, Z.; et al. Whole-genome sequencing provides insights into the genetic diversity and domestication of bitter gourd (Momordica spp.). Hortic. Res. 2020, 7, 85. [Google Scholar] [CrossRef]
- Sakio, H.; Kon, A.; Kubo, M.; Nakano, Y. Long-term fluctuations and mechanisms of seed production of riparian tree canopy species. Ecol. Res. 2023, 38, 370–385. [Google Scholar] [CrossRef]
- Li, L.; Du, Z.; Liang, J.; Mo, X.; Xu, G.; Zhu, J.; Li, H. Homogenization characteristics and regional effects in the diversity pattern of woody plants in 101 cities in China. J. Environ. Manag. 2023, 345, 118665. [Google Scholar] [CrossRef]
- Nicotra, A.B.; Beever, E.A.; Robertson, A.L.; Hofmann, G.E.; O’Leary, J. Assessing the components of adaptive capacity to improve conservation and management efforts under global change. Conserv. Biol. 2015, 29, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Plaisted, H.K.; Novak, A.B.; Weigel, S.; Klein, A.S.; Short, F.T. Eelgrass genetic diversity influences resilience to stresses associated with eutrophication. Estuaries Coasts 2020, 43, 1425–1438. [Google Scholar] [CrossRef]
- Wang, X.; Duan, F.; Zhang, H.; Han, H.; Gan, X. Fine-scale spatial genetic structure of the endangered plant Tetracentron sinense Oliv. (Trochodendraceae) in Leigong Mountain. Glob. Ecol. Conserv. 2023, 41, e02382. [Google Scholar] [CrossRef]
- Elshibli, S.; Korpelainen, H. Genetic Diversity and Population Structure of Medemia argun (Mart.) Wurttenb. ex H.Wendl. Based on Genome-Wide Markers. Front. Ecol. Evol. 2021, 9, 687188. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, Z.; Zheng, J.; Zhou, J.; Wang, B.; Zhuge, Q.; He, X. Population Genetic Diversity and Structure of an Endangered Salicaceae Species in Northeast China: Chosenia arbutifolia (Pall.). A. Skv. For. 2021, 12, 1282. [Google Scholar] [CrossRef]
- Thompson, L.; Thurman, L.; Cook, C.; Beever, E.; Sgro, C.; Battles, A.; Botero, C.; Gross, J.; Hall, K.; Hendry, A.; et al. Connecting research and practice to enhance the evolutionary potential of species under climate change. Conserv. Sci. Pract. 2023, 5, e12855. [Google Scholar] [CrossRef]
- Peláez, P.; Ortiz-Martínez, A.; Figueroa-Corona, L.; Montes, J.R.; Gernandt, D.S. Population structure, diversifying selection, and local adaptation in Pinus patula. Am. J. Bot. 2020, 107, 1555–1566. [Google Scholar] [CrossRef]
- Castillo, J.A.; Epps, C.W.; Jeffress, M.R.; Ray, C.; Rodhouse, T.J.; Schwalm, D. Replicated landscape genetic and network analyses reveal wide variation in functional connectivity for American pikas. Ecol. Appl. 2016, 26, 1660–1676. [Google Scholar] [CrossRef]
- Hu, C.L. Establishment of Molecular ID for Fraxinus Based on SSR Markers and Analysis of Genetic Diversity. Master’s Thesis, Shandong Agricultural University, Tai’an, China, 2015. [Google Scholar]
- Huff, M.; Seaman, J.; Wu, D.; Zhebentyayeva, T.; Kelly, L.J.; Faridi, N.; Nelson, C.D.; Cooper, E.; Best, T.; Steiner, K.; et al. A high-quality reference genome for Fraxinus pennsylvanica for ash species restoration and research. Mol. Ecol. Resour. 2022, 22, 1284–1302. [Google Scholar] [CrossRef]
- Meng, X.T. Study on genetic differentiation of Fraxianus mandshurica Rupr. in different provenances Northeast of China. Master’s Thesis, Northeast Forestry University, Harbin, China, 2009. [Google Scholar]
- Zheng, P.L.; Cheng, J.R.; Cheng, L.Q.; Zhou, M. Assessment of genetic diversity of an endangered species Fraxinus hupehensis based on ISSR markers. Not. Bot. Horti Agrobot. 2019, 47, 1308–1315. [Google Scholar] [CrossRef]
- Gao, C.C.; Yan, L.P.; Wu, D.J.; Wang, Y.H. Analysis of the genetic diversity and population structure of Fraxinus spp. populations based on SSR markers. J. Cent. South. Univ. For. Technol. 2023, 43, 69–78. [Google Scholar] [CrossRef]
- Jiang, T.Y.; Zhu, X.R.; Shi, C.Y.; Ye, L.Q.; Wen, G.W.; Lai, W.F.; Lv, Z.W.; Zhang, G.F. Genetic Diversity Analysis of Fraxinus mandshurica Based on dd-RAD Simplified Genome Sequencing. Mol. Plant Breed 2022, 1–16. [Google Scholar]
- Jing, D.; Liu, X.; He, Q.; Dang, J.; Hu, R.; Xia, Y.; Wu, D.; Wang, S.; Zhang, Y.; Xia, Q.; et al. Genome assembly of wild loquat (Eriobotrya japonica) and resequencing provide new insights into the genomic evolution and fruit domestication in loquat. Hortic. Res. 2022, 10, 265. [Google Scholar] [CrossRef]
- Mekbib, Y.; Tesfaye, K.; Dong, X.; Saina, J.K.; Hu, G.W.; Wang, Q.F. Whole-genome resequencing of Coffea arabica L. (Rubiaceae) genotypes identify SNP and unravels distinct groups showing a strong geographical pattern. BMC Plant Biol. 2022, 22, 69. [Google Scholar] [CrossRef]
- Qing, J.; Meng, Y.D.; He, F.; Du, Q.X.; Zhong, J.; Du, H.Y.; Liu, P.F.; Du, L.Y.; Wang, L. Whole genome re-sequencing reveals the genetic diversity and evolutionary patterns of Eucommia ulmoides. Mol. Genet. Genom. 2022, 297, 485–494. [Google Scholar] [CrossRef]
- Tel-Zur, N.; Abbo, S.; Myslabodski, D.; Mizrahi, Y. Modified CTAB Procedure for DNA Isolation from Epiphytic cacti of the Genera Hylocereus and Selenicereus (Cactaceae). Plant Mol. Biol. Rep. 1999, 17, 249–254. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- do Valle, Í.F.; Giampieri, E.; Simonetti, G.; Padella, A.; Manfrini, M.; Ferrari, A.; Papayannidis, C.; Zironi, I.; Garonzi, M.; Bernardi, S.; et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinform. 2016, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Langella, O.; Chikhi, L.; Beaumont, M.A. LEA (likelihood-based estimation of admixture): A program to simultaneously estimate admixture and the time since admixture. Mol. Ecol. Resour. 2010, 1, 357–358. [Google Scholar] [CrossRef]
- Zhou, Y.; Qiu, H.; Xu, S. Modeling Continuous Admixture Using Admixture-Induced Linkage Disequilibrium. Sci. Rep. 2017, 7, 43054. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2010, 88, 76–82. [Google Scholar] [CrossRef]
- Privé, F.; Luu, K.; Blum, M.G.B.; McGrath, J.J.; Vilhjálmsson, B.J. Efficient toolkit implementing best practices for principal component analysis of population genetic data. Bioinformatics 2020, 36, 4449–4457. [Google Scholar] [CrossRef]
- Garreta, L.; Cerón-Souza, I.; Palacio, M.R.; Reyes-Herrera, P.H. Multi GWAS: An integrative tool for Genome Wide Association Studies in tetraploid organisms. Ecol. Evol. 2021, 11, 7411–7426. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, F.; Zhao, S.; Bo, W.; Jiang, L.; Pang, X.; Wu, R. Inferring the evolutionary history of outcrossing populations through computing a multiallelic linkage–linkage disequilibrium map. Methods Ecol. Evol. 2015, 6, 1259–1269. [Google Scholar] [CrossRef]
- Campoy, J.A.; Lerigoleur-Balsemin, E.; Christmann, H.; Beauvieux, R.; Girollet, N.; Quero-García, J.; Dirlewanger, E.; Barreneche, T. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection of Prunus avium L. landraces and bred cultivars. BMC Plant Biol. 2016, 16, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Guillot, G.; Rousset, F. Dismantling the mantel tests. Methods Ecol. Evol. 2013, 4, 336–344. [Google Scholar] [CrossRef]
- Tian, Q.; El-Kassaby, Y.A.; Li, W. Revealing the Genetic Structure and Differentiation in Endangered Pinus bungeana by Genome-Wide SNP Markers. Forests 2022, 13, 326. [Google Scholar] [CrossRef]
- Linck, E.; Battey, C.J. Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Mol. Ecol. Resour. 2019, 19, 639–647. [Google Scholar] [CrossRef]
- Shete, S.; Tiwari, H.; Elston, R.C. On estimating the heterozygosity and polymorphism information content value. Theor. Popul. Biol. 2000, 57, 265–271. [Google Scholar] [CrossRef]
- José, G.D.; Akanksha, S.; Corbinian, G.; Stetter, M.G. Genetic Incompatibilities and Evolutionary Rescue by Wild Relatives Shaped Grain Amaranth Domestication. Mol. Biol. Evol. 2023, 40, msad177. [Google Scholar] [CrossRef]
- Ibañez, E.A.; Guichón, M.L.; Peralta, D.M.; Cassini, M.H.; Túnez, J.I. Genetic structure and gene flow in Myocastor coypus (Rodentia: Echimyidae) populations inhabiting a highly modified agroecosystem of the Pampas region. Biol. J. Linn. Soc. 2024, 141, 401–418. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, F.Y.; Zhang, H.X. Evolutionary Genomics Provides Insights Into Endangerment and Conservation of a Wild Apple Tree Species, Malus sieversii. Evol. Appl. 2024, 17, e70048. [Google Scholar] [CrossRef]
- Yan, L.P.; Wu, D.J.; Wang, Y.H.; Liu, C.; Wang, K.; Wu, H.; Li, Q. Analysis on phylogenetic relationship of elite germplasm resources in Fraxinus using AFLP. J. Cent. South. Univ. For. Technol. 2018, 38, 89–95. [Google Scholar] [CrossRef]
- Hu, L.J.; Uchiyama, K.; Shen, H.L.; Saito, Y.; Tsuda, Y.; Ide, Y. Nuclear DNA microsatellites reveal genetic variation but a lack of phylogeographical structure in an endangered species, Fraxinus mandshurica, across North-east China. Ann. Bot. 2008, 102, 195–205. [Google Scholar] [CrossRef]
- Sagarin, R.D.; Gaines, S.D. The ‘abundant centre’ distribution: To what extent is it a biogeographical rule? Ecol. Lett. 2002, 5, 137–147. [Google Scholar] [CrossRef]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic variation across species’ geographical ranges: The central-marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Pironon, S.; Papuga, G.; Villellas, J.; Angert, A.L.; García, M.B.; Thompson, J.D. Geographic variation in genetic and demographic performance: New insights from an old biogeographical paradigm. Biol. Rev. 2017, 92, 1877–1909. [Google Scholar] [CrossRef]
- Abhainn, E.A.; Shirley, D.L.; Stanley, R.K.; Scarpato, T.; Koch, J.L.; Romero-Severson, J. Gene flow from Fraxinus cultivars into natural stands of Fraxinus pennsylvanica occurs range-wide, is regionally extensive, and is associated with a loss of allele richness. PLoS ONE 2024, 19, e0294829. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | MAF | PIC | Pi | Ho | He | Tajima’s D |
|---|---|---|---|---|---|---|
| 1 | 0.0722 (6.0313 × 10−5) | 0.2190 (7.6915 × 10−5) | 0.0005 (3 × 10−7) | 0.1191 (6.8796 × 10−3) | 0.1082 (4.1688 × 10−5) | −0.1005 (4.20 × 10−3) |
| 2 | 0.0707 (6.3403 × 10−5) | 0.2410 (7.5970 × 10−5) | 0.0005 (3 × 10−7) | 0.117 (7.3985 × 10−3) | 0.1031 (4.9169 × 10−5) | −0.1983 (4.10 × 10−3) |
| 3 | 0.2609 (9.2414 × 10−5) | 0.3198 (4.5299 × 10−5) | 0.0016 (7 × 10−7) | 0.1081 (8.7903 × 10−3) | 0.3297 (2.2090 × 10−4) | 1.4255 (4.90 × 10−3) |
| 4 | 0.0674 (7.4947 × 10−5) | 0.3224 (3.5787 × 10−5) | 0.0005 (3 × 10−7) | 0.1197 (1.0726 × 10−2) | 0.0866 (1.8200 × 10−4) | −0.058 (3.30 × 10−3) |
| 5 | 0.1385 (5.0368 × 10−5) | 0.2083 (3.8242 × 10−5) | 0.0011 (4 × 10−7) | 0.1213 (6.9485 × 10−3) | 0.2213 (8.3344 × 10−5) | −0.6303 (2.00 × 10−3) |
| 6 | 0.0805 (6.8972 × 10−5) | 0.2669 (6.3191 × 10−5) | 0.0006 (3 × 10−7) | 0.1313 (1.6690 × 10−2) | 0.1155 (6.2816 × 10−5) | −0.186 (3.60 × 10−3) |
| 7 | 0.0783 (6.4654 × 10−5) | 0.2373 (7.3850 × 10−5) | 0.0006 (3 × 10−7) | 0.1367 (8.0000 × 10−3) | 0.1157 (3.8717 × 10−5) | −0.0972 (3.90 × 10−3) |
| 8 | 0.1210 (6.6023 × 10−5) | 0.2597 (4.2896 × 10−5) | 0.0009 (4 × 10−7) | 0.1098 (3.4593 × 10−2) | 0.1837 (5.5073 × 10−5) | −0.0947 (2.50 × 10−3) |
| 9 | 0.0718 (6.2211 × 10−5) | 0.2373 (7.2699 × 10−5) | 0.0005 (3 × 10−7) | 0.1167 (1.2470 × 10−4) | 0.1058 (2.0041 × 10−2) | −0.2806 (3.90 × 10−3) |
| 10 | 0.0707 (6.7828 × 10−5) | 0.2718 (6.9322 × 10−5) | 0.0005 (3 × 10−7) | 0.1239 (3.2038 × 10−2) | 0.0992 (2.0313 × 10−4) | −0.0604 (4.10 × 10−3) |
| 11 | 0.2481 (7.8788 × 10−5) | 0.2923 (5.0968 × 10−5) | 0.0016 (6 × 10−7) | 0.1177 (4.8836 × 10−3) | 0.3343 (3.8793 × 10−4) | 1.3141 (5.60 × 10−3) |
| Group | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 0.0276 | |||||||||
| 3 | 0.4560 | 0.3986 | ||||||||
| 4 | 0.0170 | 0.0269 | 0.2240 | |||||||
| 5 | 0.0395 | 0.0195 | 0.2140 | −0.1119 | ||||||
| 6 | 0.0198 | 0.0262 | 0.3307 | −0.0117 | −0.0483 | |||||
| 7 | 0.0210 | 0.0336 | 0.4103 | 0.0150 | 0.0224 | −0.0041 | ||||
| 8 | 0.0562 | 0.0354 | 0.2160 | −0.0982 | −0.0496 | −0.0128 | 0.0375 | |||
| 9 | 0.0143 | 0.0223 | 0.3920 | −0.0221 | 0.0058 | 0.0033 | 0.0125 | 0.0193 | ||
| 10 | 0.0353 | 0.0450 | 0.3468 | 0.0328 | −0.0093 | 0.0242 | 0.0349 | 0.0074 | 0.0222 | |
| 11 | 0.2049 | 0.1588 | −0.0484 | −0.0092 | 0.0255 | 0.1026 | 0.1690 | 0.0200 | 0.1489 | 0.1156 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Ge, W.; Zhao, Q.; Zhang, J.; Guo, X.; Ma, W. Genome-Wide Resequencing Revealed the Genetic Diversity of Fraxinus platypoda Oliv. in Northwestern China. Forests 2025, 16, 860. https://doi.org/10.3390/f16050860
Liu Y, Ge W, Zhao Q, Zhang J, Guo X, Ma W. Genome-Wide Resequencing Revealed the Genetic Diversity of Fraxinus platypoda Oliv. in Northwestern China. Forests. 2025; 16(5):860. https://doi.org/10.3390/f16050860
Chicago/Turabian StyleLiu, Ying, Wanting Ge, Qiuling Zhao, Jing Zhang, Xiaolong Guo, and Wenjun Ma. 2025. "Genome-Wide Resequencing Revealed the Genetic Diversity of Fraxinus platypoda Oliv. in Northwestern China" Forests 16, no. 5: 860. https://doi.org/10.3390/f16050860
APA StyleLiu, Y., Ge, W., Zhao, Q., Zhang, J., Guo, X., & Ma, W. (2025). Genome-Wide Resequencing Revealed the Genetic Diversity of Fraxinus platypoda Oliv. in Northwestern China. Forests, 16(5), 860. https://doi.org/10.3390/f16050860