The Phyllosphere Microbial Community Structure of Three Camellia Species upon Anthracnose


Abstract
1. Introduction
2. Methods
2.1. Sample Collection
2.2. Microbial DNA Extraction, PCR Amplification, and Illumina Sequencing
2.3. Bioinformatics Analysis
2.4. Isolation and Culture of Microorganisms from Anthracnose Lesions
2.5. Co-Culture and Tie-Back Experiment
3. Results
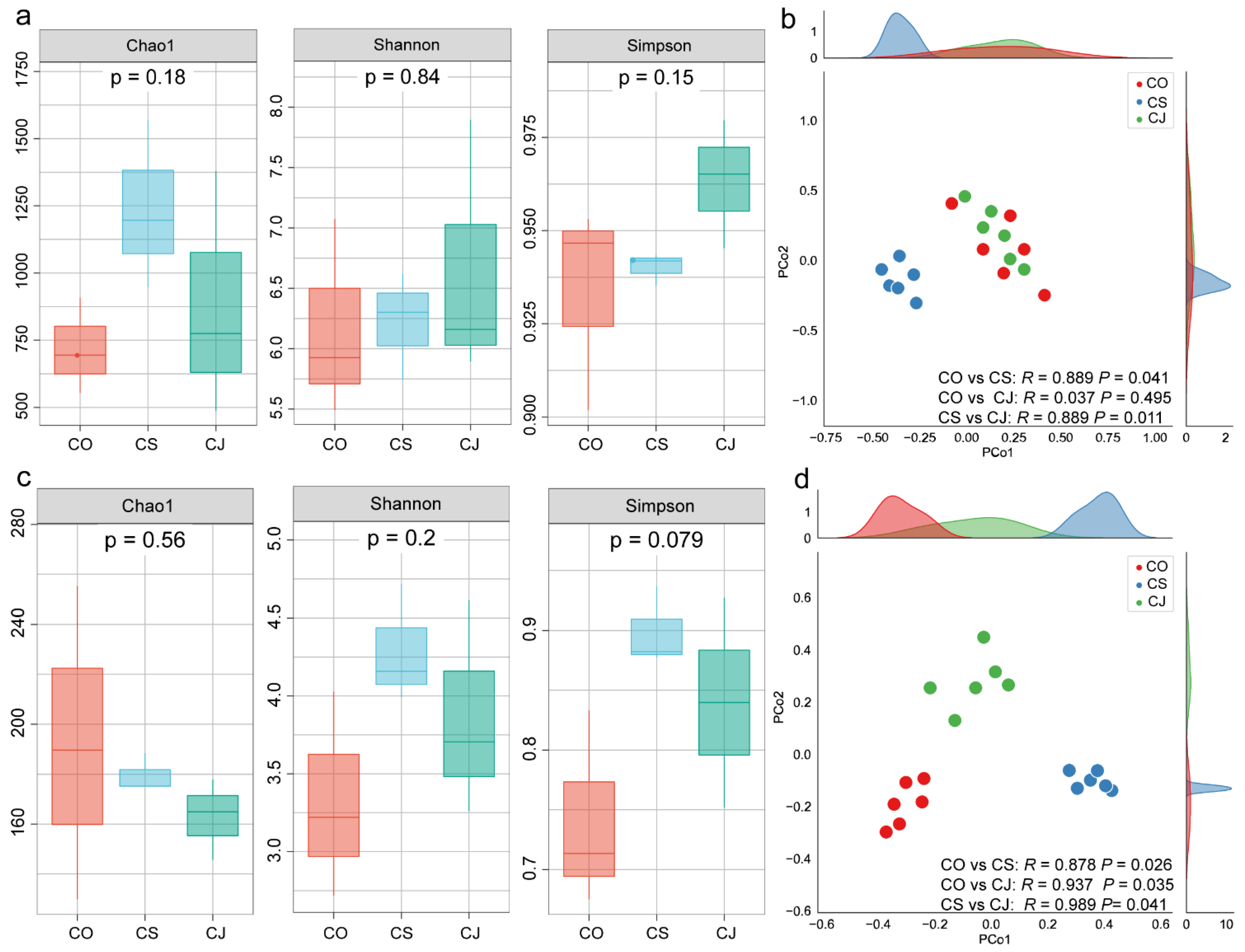
3.1. Phyllosphere Fungal and Bacterial Community Diversity of Three Camellia Plants Under Anthracnose
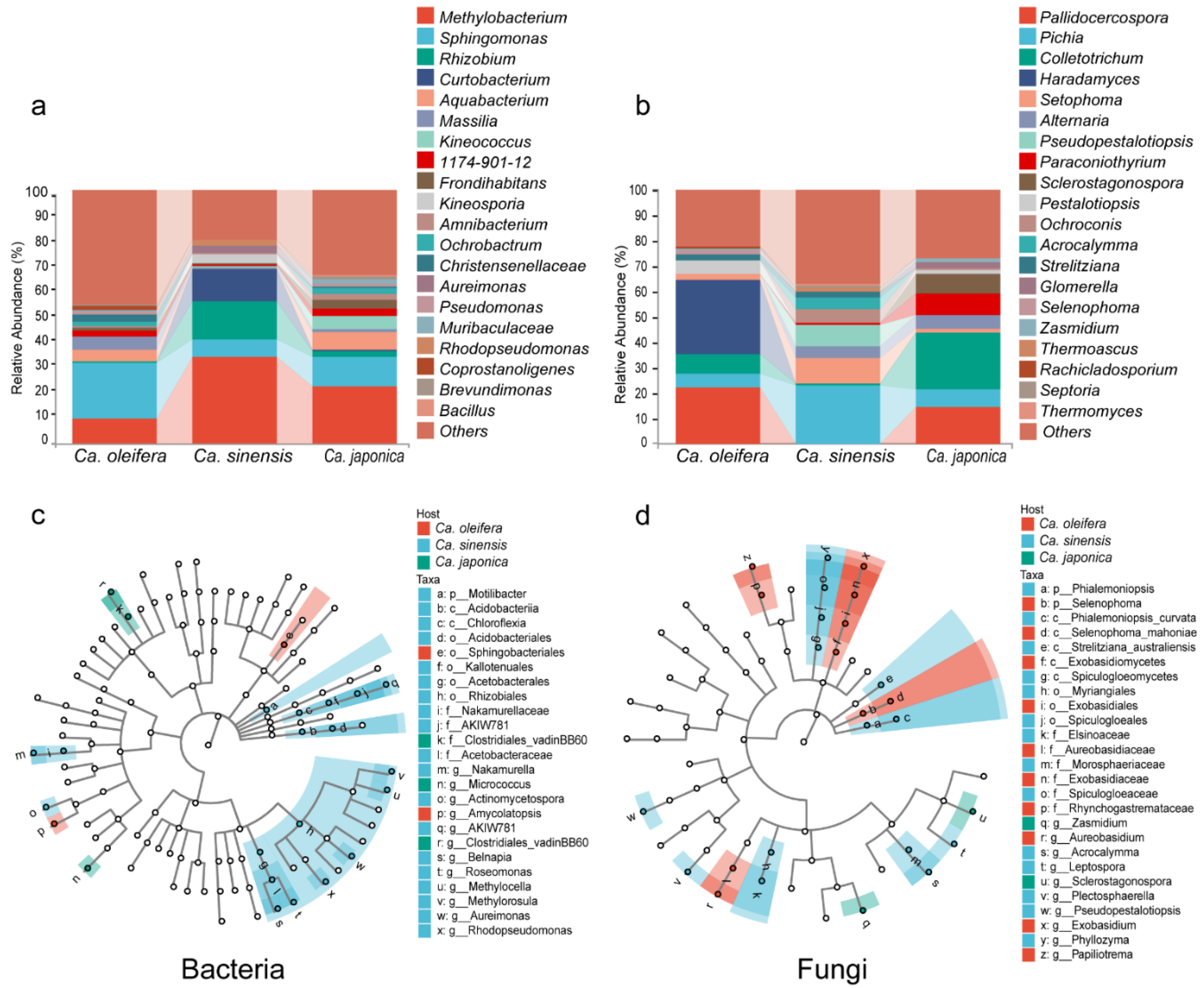
3.2. Phyllosphere Bacterial and Fungal Community Composition of Three Camellia Plants Under Anthracnose
3.3. Differential Abundance of the Phyllosphere Bacterial and Fungal Taxa Among Three Camellia Plants
3.4. Phyllosphere Key Microbial Taxa and Their Associations with Three Camellia Plants Under Anthracnose
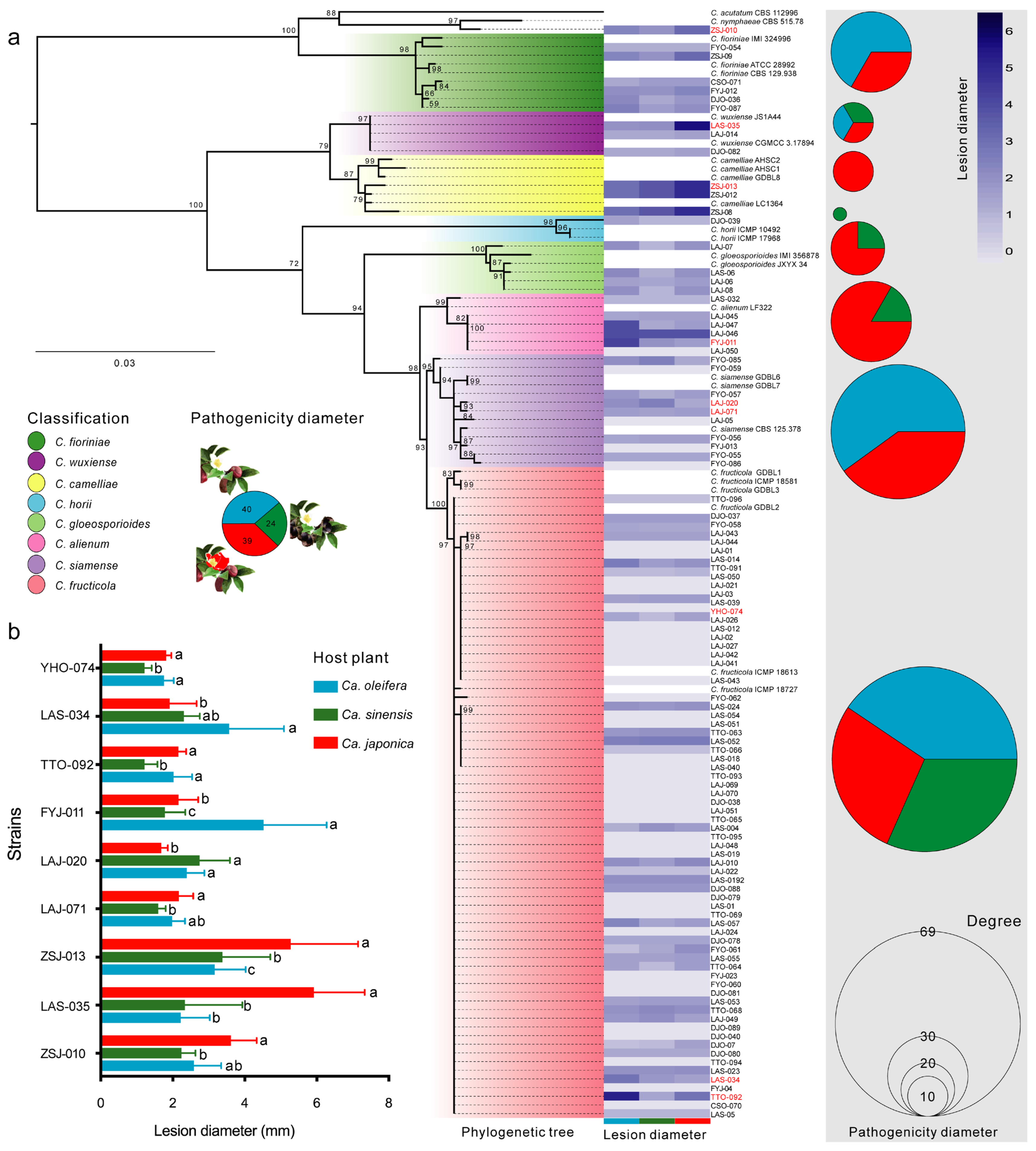
3.5. Microorganisms Associated with the Three Camellia Phyllosphere Anthracnose Lesion Microbiome
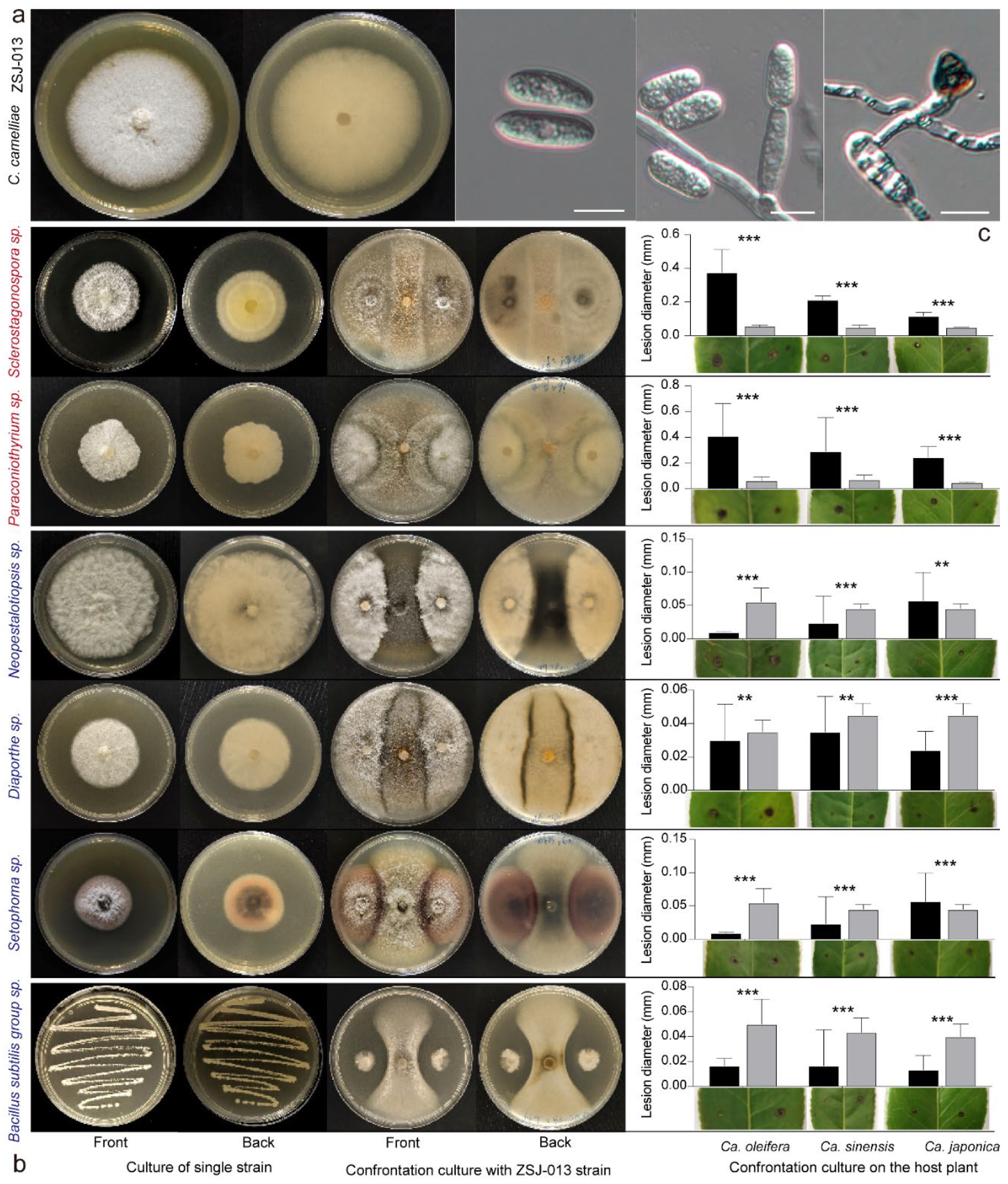
3.6. Members of the Phyllosphere Microbiome Could Affect Colletotrichum Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, P.T.; Whiteman, N.K. Insect herbivory reshapes a native leaf microbiome. Nat. Ecol. Evol. 2020, 4, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Zhang, X.Y.; Zhang, Z.C.; Chen, Y.; Tian, Q.; Zeng, D.D.; Xu, M.; Wang, Y.; Dong, S.M.; Ma, Z.H.; et al. Fusarium–produced vitamin B6 promotes the evasion of soybean resistance by Phytophthora sojae. J. Integr. Plant Biol. 2023, 65, 2204–2217. [Google Scholar] [CrossRef]
- Yin, C.; Casa Vargas, J.M.; Schlatter, D.C.; Hagerty, C.H.; Hulbert, S.H.; Paulitz, T.C. Rhizosphere community selection reveals bacteria associated with reduced root disease. Microbiome 2021, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ameye, M.; Landschoot, S.; De Zutter, N.; De Saeger, S.; De Boevre, M.; Audenaert, K. At the scene of the crime: New insights into the role of weakly pathogenic members of the fusarium head blight disease complex. Mol. Plant Pathol. 2020, 21, 1559–1572. [Google Scholar] [CrossRef]
- Espinoza, J.G.; Briceo, E.X.; Keith, L.M.; Latorre, B.A. Canker and twig dieback of blueberry caused by Pestalotiopsis spp. and a Truncatella sp. in Chile. Plant Dis. 2008, 92, 1375–1475. [Google Scholar] [CrossRef]
- Shi, Y.L. Studies on Molecular Mechanism of Tea Plant Against Anthracnose Infection and Assessment of Freezing Tolerance of Tea Cultivars. Ph.D. Dissertation, Zhejiang University, Hangzhou, China, 2020. [Google Scholar]
- Zhang, Z.; Luo, L.; Tan, X.; Kong, X.; Yang, J.; Wang, D.; Zhang, D.; Jin, D.; Liu, Y. Pumpkin powdery mildew disease severity influences the fungal diversity of the phyllosphere. Peerj 2018, 6, e4559. [Google Scholar] [CrossRef]
- Chaudhry, V.; Runge, P.; Sengupta, P.; Doehlemann, G.; Parker, J.E.; Kemen, E. Shaping the leaf microbiota: Plant–microbe–microbe interactions. J. Exp. Bot. 2021, 72, 36–56. [Google Scholar] [CrossRef]
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Microbiol. 2012, 10, 828–840. [Google Scholar] [CrossRef]
- Gong, T.Y.; Xin, X.F. Phyllosphere microbiota: Community dynamics and its interaction with plant hosts. J. Integr. Plant Biol. 2021, 63, 297–304. [Google Scholar] [CrossRef]
- Fan, X.; Matsumoto, H.; Xu, H.; Fang, H.; Pan, Q.; Lv, T.; Zhan, C.; Feng, X.; Liu, X.; Su, D.; et al. Aspergillus cvjetkovicii protects against phytopathogens through interspecies chemical signalling in the phyllosphere. Nat. Microbiol. 2024, 9, 2862–2876. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.M.; Sousa, C. A Review on the Biological Activity of Camellia Species. Molecules 2021, 26, 2178. [Google Scholar] [CrossRef]
- Fu, M.; Crous, P.W.; Bai, Q.; Zhang, P.F.; Xiang, J.; Guo, Y.S.; Zhao, F.F.; Yang, M.M.; Hong, N.; Xu, W.X.; et al. Colletotrichum species associated with anthracnose of Pyrus spp. in China. Persoonia 2019, 42, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, V.; Groenewald, J.Z.; Polizzi, G.; Crous, P.W. High species diversity in Colletotrichum associated with citrus diseases in Europe. Persoonia 2018, 39, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.R.; Xu, X.M.; Che, H.Y.; West, J.S.; Luo, D.Q. Characteristics and distribution of Colletotrichum species in coffee plantations in Hainan, China. Plant Pathol. 2019, 68, 1146–1156. [Google Scholar] [CrossRef]
- Peng, X.J.; Wang, Q.C.; Zhang, S.K.; Guo, K.; Zhou, X.D. Colletotrichum species associated with Camellia anthracnose in China. Mycosphere 2023, 14, 130–157. [Google Scholar] [CrossRef]
- Wang, Y.C.; Hao, X.Y.; Wang, L.; Xiao, B.; Wang, X.C.; Yang, Y.J. Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep. 2016, 6, 35287. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Cao, H.; Hao, X.; Zeng, J.; Yang, Y.; Wang, X. Transcriptome analysis of an anthracnose-resistant tea plant cultivar reveals genes associated with resistance to Colletotrichum camelliae. PLoS ONE 2017, 11, e0148535. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High–resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson–Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence–based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open–source, platform–independent, community–supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Chang, F.; He, S.; Dang, C. Assisted Selection of Biomarkers by Linear Discriminant Analysis Effect Size (LEfSe) in Microbiome Data. J. Vis. Exp. 2022, 11, 537–558. [Google Scholar] [CrossRef]
- Parente, E.; Cocolin, L.; De Filippis, F.; Zotta, T.; Ferrocino, I.; O’Sullivan, O.; Neviani, E.; De Angelis, M.; Cotter, P.D.; Ercolini, D. FoodMicrobionet: A database for the visualisation and exploration of food bacterial communities based on network analysis. Int. J. Food Microbiol. 2016, 219, 28–37. [Google Scholar] [CrossRef]
- White, T.; Bruns, T.; Lee, S.; Taylor, F.J.R.M.; White, T.; Lee, S.H.; Taylor, L.; Shawetaylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. 1990, 38, 315–322. [Google Scholar] [CrossRef]
- Carbone, I.; Kohn, L.M. A Method for Designing Primer Sets for Speciation Studies in Filamentous Ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- O’Donnell, K.; Cigelnik, E. Two divergent intragenomic rDNAITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol. Phylogenet. Evol. 1997, 7, 103–116. [Google Scholar] [CrossRef]
- Templeton, M.D.; Rikkerink, E.H.; Solon, S.L.; Crowhurst, R.N. Cloning and molecular characterization of the glyceraldehyde-3-phosphate dehydrogenase-encoding gene and cDNA from the plant pathogenic fungus Glomerella cingulata. Gene 1992, 122, 225. [Google Scholar] [CrossRef]
- Weir, B.S.; Johnston, P.R.; Damm, U. The Colletotrichum gloeosporioides species complex. Stud. Mycol. 2012, 73, 115–180. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.M.; Zhou, X.; Zhang, A.M.; Cai, L. Disease–induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef]
- Li, P.D.; Zhu, Z.R.; Zhang, Y.; Xu, J.; Wang, H.; Wang, Z.; Li, H. The phyllosphere microbiome shifts toward combating melanose pathogen. Microbiome 2022, 10, 56. [Google Scholar] [CrossRef]
- Aziz, M.Z.; Yaseen, M.; Abbas, T.; Naveed, M.; Mustafa, A.; Hamid, Y.; Saeed, Q.; Ming-Gang, X. Foliar application of micronutrients enhances crop stand, yield and the biofortification essential for human health of different wheat cultivars. J. Integr. Agric. 2019, 18, 1369–1378. [Google Scholar] [CrossRef]
- Sy, A.; Timmers, A.C.; Knief, C.; Vorholt, J.A. Methylotrophic metabolism is advantageous for Methylobacterium extorquens during colonization of Medicago truncatula under competitive conditions. Appl. Environ. Microbiol. 2005, 71, 7245–7252. [Google Scholar] [CrossRef]
- Knief, C.; Frances, L.; Vorholt, J.A. Competitiveness of Diverse Methylobacterium Strains in the Phyllosphere of Arabidopsis thaliana and Identification of Representative Models, Including M. extorquens PA1. Microb. Ecol. 2010, 60, 440–452. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, M.Y.; Khan, N.; Tan, L.L.; Yang, S. Potentials, Utilization, and Bioengineering of Plant Growth–Promoting Methylobacterium for Sustainable Agriculture. Sustainability 2021, 13, 3941. [Google Scholar] [CrossRef]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- Sedláček, I.; Holochová, P.; Busse, H.J.; Koublová, V.; Králová, S.; Švec, P.; Sobotka, R.; Staňková, E.; Pilný, J.; Šedo, O.; et al. Characterisation of Waterborne Psychrophilic Massilia Isolates with Violacein Production and Description of Massilia antarctica sp. nov. Microorganisms 2022, 10, 704. [Google Scholar] [CrossRef]
- Kanitkar, S.; Sawant, S.; Adsule, P.; Kulkarni, M.; Kadam, M.; Raut, V. Bio-Efficacy of Milastin–K (Bacillus subtilis KTSB 1015 1.5% A.S.) as a Potential Bio–Control Agent for Management of Bacterial Blight (Xanthomonas axonopodis) and Anthracnose (Colletotrichum gloeosporioides) Diseases in Pomegranate. Soc. Sci. Electron. Publ. 2020, 7, 2349–8889. [Google Scholar] [CrossRef]
- Wang, X.; Yuan, Z.; Shi, Y.; Cai, F.; Wang, Y. Bacillus amyloliquefaciens HG01 induces resistance in loquats against anthracnose rot caused by Colletotrichum acutatum. Postharvest Biol. Technol. 2020, 160, 111034. [Google Scholar] [CrossRef]
- Huang, L.; Li, Q.C.; Hou, Y.; Li, G.Q.; Ye, J.R. Bacillus velezensis strain HYEB5-6 as a potential biocontrol agent against anthracnose on Euonymus japonicus. Biocontrol. Sci. Technol. 2017, 27, 636–653. [Google Scholar] [CrossRef]
- Bashir, I.; War, A.F.; Rafiq, I.; Reshi, Z.A.; Rashid, I.; Shouche, Y.S. Phyllosphere microbiome: Diversity and functions. Microbiol. Res. 2022, 254, 126888. [Google Scholar] [CrossRef]
- López-López, A.M.; León-Félix, J.; Allende, R.; Lima, N.B.; García–Estrada, R.S. First Report of Setophoma terrestris Causing Corky and Pink Root of Tomato in Sinaloa, Mexico. Plant Dis. 2020, 104, 1553. [Google Scholar] [CrossRef]
- Zhang, F.B.; Zheng, H.L.; Cui, W.G.; Zhang, M.Q.; Yin, Y.S.; Cui, M.; Gao, M. First Report of Setophoma terrestris Causing Pink Root of Garlic in China. Plant Dis. 2019, 103, 584. [Google Scholar] [CrossRef]
- Ikeda, K.; Kuwabara, K.; Urushibara, T.; Soyai, P.; Miki, S.; Shibata, S. Pink root rot of squash caused by Setophoma terrestris in Japan. J. Gen. Plant Pathol. 2012, 78, 372–375. [Google Scholar] [CrossRef]
- Yoshida, N. Seasonal dynamics of the pink root fungus (Setophoma terrestris) in rhizosphere soil: Effect of crop species and rotation. Plant Pathol. 2022, 71, 361–372. [Google Scholar] [CrossRef]
- Feau, N.; Weiland, J.E.; Stanosz, G.R.; Bernier, L. Specific and sensitive PCR–based detection of Septoria musiva, S. populicola and S. populi, the causes of leaf spot and stem canker on poplars. Mycol. Res. 2005, 109, 1015–1028. [Google Scholar] [CrossRef]
- Satelis, F.J.; Boiteux, S.L.; Reis, A. Resistance to Septoria lycopersici in Solanum (section Lycopersicon) species and in progenies of S. lycopersicum × S. peruvianum. Sci. Agric. 2010, 67, 334–341. [Google Scholar] [CrossRef]
- Forootanfar, H.; Movahednia, M.M.; Yaghmaei, S.; Tabatabaei-Sameni, M.; Rastegar, H.; Sadighi, A.; Faramarzi, M.A. Removal of chlorophenolic derivatives by soil isolated ascomycete of Paraconiothyrium variabile and studying the role of its extracellular laccase. J. Hazard. Mater. 2012, 209, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Montecchio, L.; Causin, R.; Vettorazzo, M. A Twig Canker on English Hawthorn Caused by Coniothyrium sporulosum in Italy. Plant Dis. 2002, 86, 1403. [Google Scholar] [CrossRef]
- Cloete, M.; Fourie, P.H.; Damm, U.; Crous, P.W.; Mostert, L. Fungi associated with die-back symptoms of apple and pear trees, a possible inoculum source of grapevine trunk disease pathogens. Phytopathol. Mediterr. 2011, 50, 176–190. [Google Scholar]
- Gao, Y.; Liu, F.; Cai, L. Unravelling Diaporthe species associated with Camellia. Syst. Biodivers. 2016, 14, 102–117. [Google Scholar] [CrossRef]
- Gai, Y.; Xiong, T.; Xiao, X.; Li, P.; Zeng, Y.; Li, L.; Riely, B.K.; Li, H. The Genome Sequence of the Citrus Melanose Pathogen Diaporthe citri and Two Citrus–Related Diaporthe Species. Phytopathology 2021, 111, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, A.J.; Liu, M.; Zhang, W.; Chen, Z.; Udayanga, D.; Chukeatirote, E.; Li, X.; Yan, J.; Hyde, K.D. Morphological and molecular characterisation of Diaporthe species associated with grapevine trunk disease in China. Fungal Biol. 2015, 119, 283–294. [Google Scholar] [CrossRef]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Primer Sequence (5′-3′) | Reference |
|---|---|---|---|
| ITS | ITS1 | TCCGTAGGTGAACCTGCGG | [28] |
| ITS4 | TCCTCCGCTTATTGATATGC | ||
| ACT | ACT-512F | ATGTGCAAGGCCGGTTTCGC | [29] |
| ACT-783R | TACGAGTCCTTCTGGCCCAT | ||
| TUB2 | T1 | AACATGCGTGAGATTGTAAGT | [30] |
| Bt2b | ACCCTCAGTGTAGTGACCCTTGGC | ||
| GAPDH | GDF | GCCGTCAACGACCCCTTCATTGA | [31] |
| GDR | GGGTGGAGTCGTACTTGAGCATGT |
| Species | Node | Positive Edge | Negative Edge | Average Degree | Modularity a | Average Clustering Coefficient b | Average Path Distance c | |
|---|---|---|---|---|---|---|---|---|
| CO | 16s | 62 | 594 | 495 | 35.129 | 9.785 | 0.871 | 1.569 |
| ITS | 41 | 265 | 166 | 21.024 | 1.957 | 0.839 | 1.633 | |
| 16s + ITS | 103 | 1485 | 1285 | 53.786 | 6.93 | 0.863 | 1.609 | |
| CS | 16s | 53 | 417 | 398 | 30.755 | 20.034 | 0.895 | 1.507 |
| ITS | 52 | 478 | 383 | 33.115 | 8.072 | 0.916 | 1.548 | |
| 16s + ITS | 105 | 1639 | 1465 | 59.124 | 17.76 | 0.884 | 1.559 | |
| CJ | 16s | 66 | 584 | 511 | 33.182 | 4.749 | 0.871 | 1.651 |
| ITS | 53 | 436 | 281 | 27.057 | 1.861 | 0.784 | 1.674 | |
| 16s + ITS | 119 | 1921 | 1560 | 58.504 | 3.272 | 0.823 | 1.662 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, X.; Wang, H.; Zhou, X. The Phyllosphere Microbial Community Structure of Three Camellia Species upon Anthracnose. Forests 2024, 15, 2080. https://doi.org/10.3390/f15122080
Peng X, Wang H, Zhou X. The Phyllosphere Microbial Community Structure of Three Camellia Species upon Anthracnose. Forests. 2024; 15(12):2080. https://doi.org/10.3390/f15122080
Chicago/Turabian StylePeng, Xiaojie, Haonan Wang, and Xudong Zhou. 2024. "The Phyllosphere Microbial Community Structure of Three Camellia Species upon Anthracnose" Forests 15, no. 12: 2080. https://doi.org/10.3390/f15122080
APA StylePeng, X., Wang, H., & Zhou, X. (2024). The Phyllosphere Microbial Community Structure of Three Camellia Species upon Anthracnose. Forests, 15(12), 2080. https://doi.org/10.3390/f15122080