
Weighted Gene Co-Expression Network Analysis Reveals Key Pathways and Hub Genes Associated with Successful Grafting in Pecan (Carya illinoinensis)
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA-seq
2.2. Selection of Variable Genes
2.3. Construction of Co-Expression Network
2.4. Identification of Biologically Significant Modules
2.5. Gene enrichment Analysis on Meaningful Modules
2.6. Screen of Hub Gene
2.7. Detecting the Promoter Variations in Hub Genes
3. Results
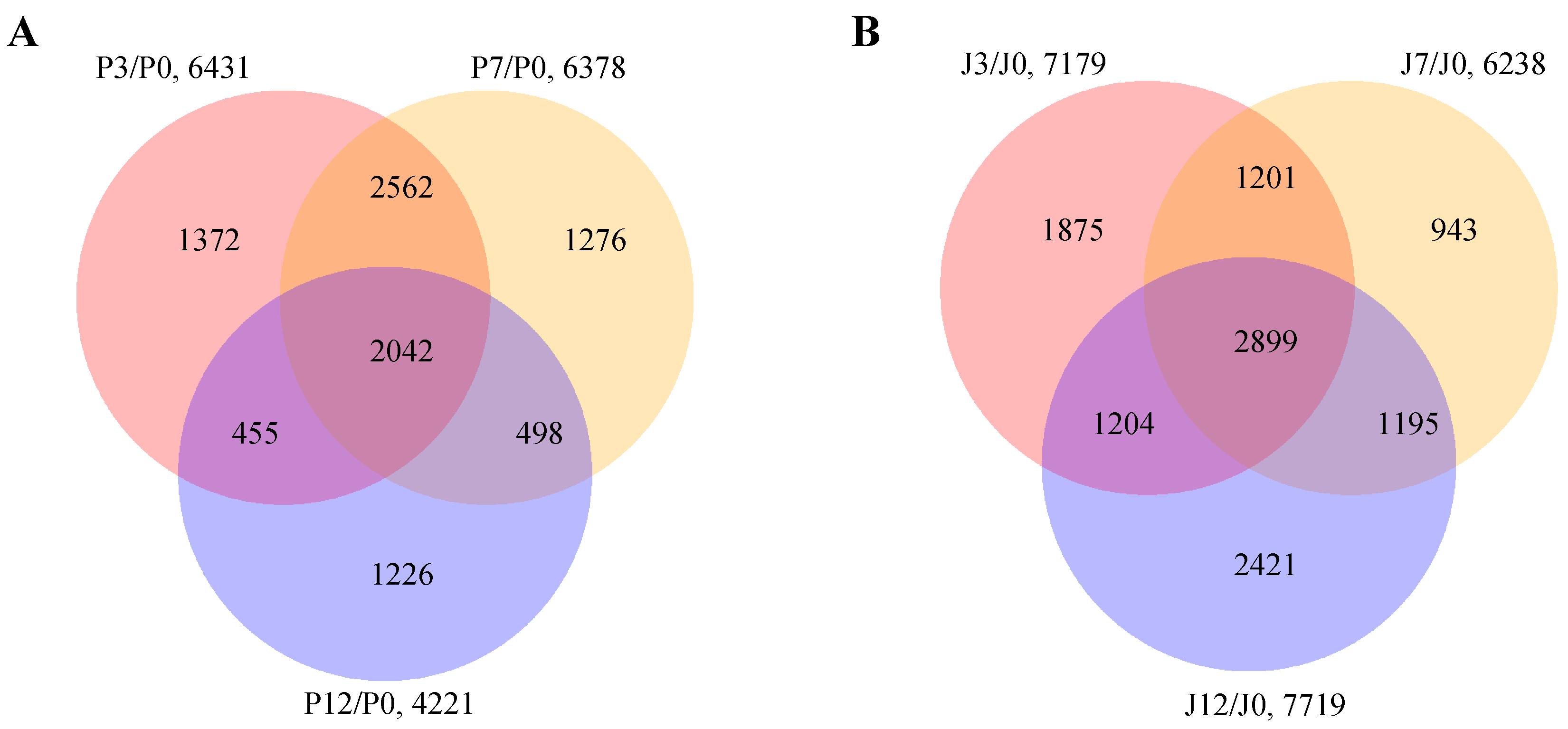
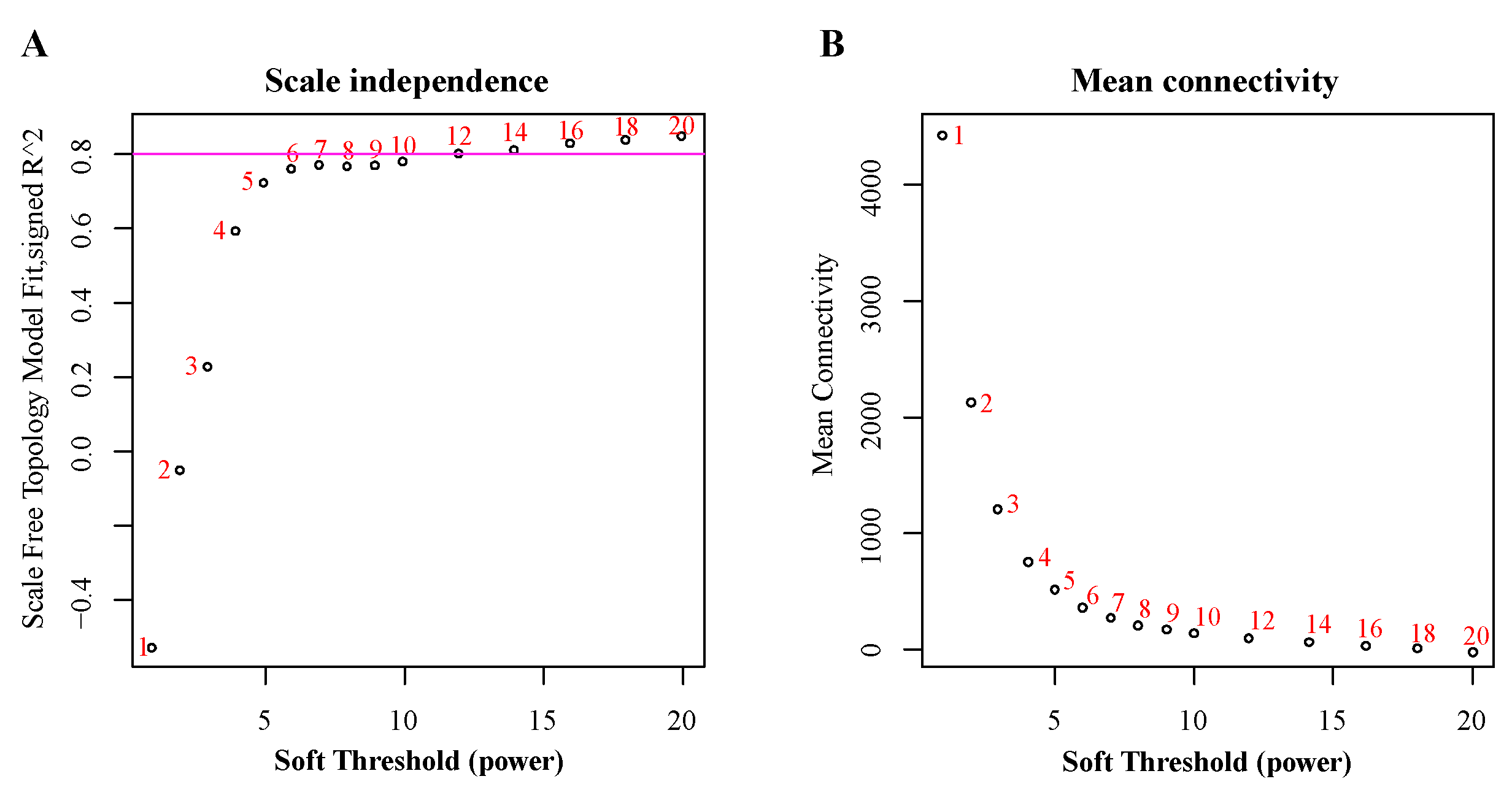
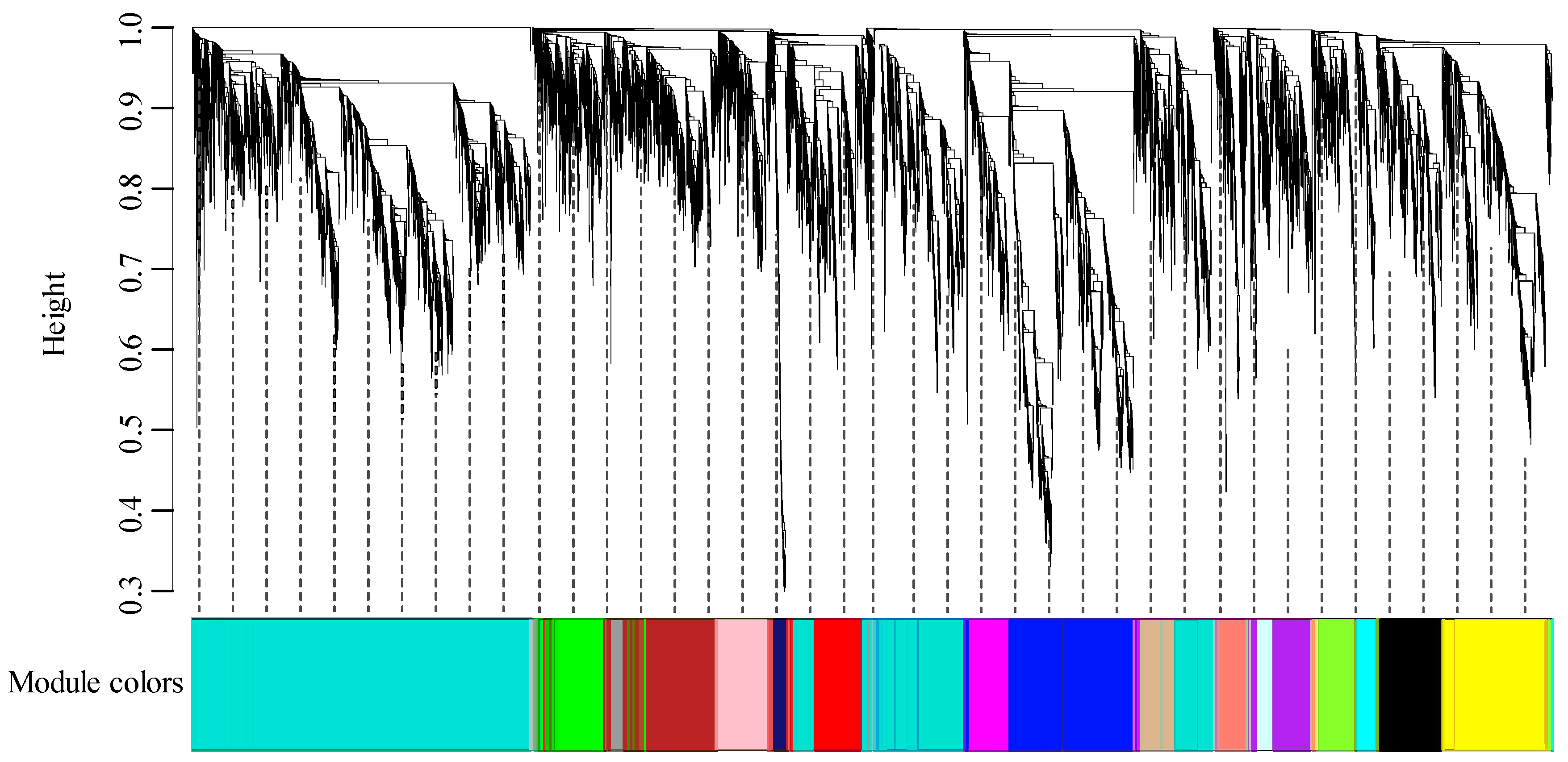
3.1. RNA Sequencing and Construction of Co-Expression Network
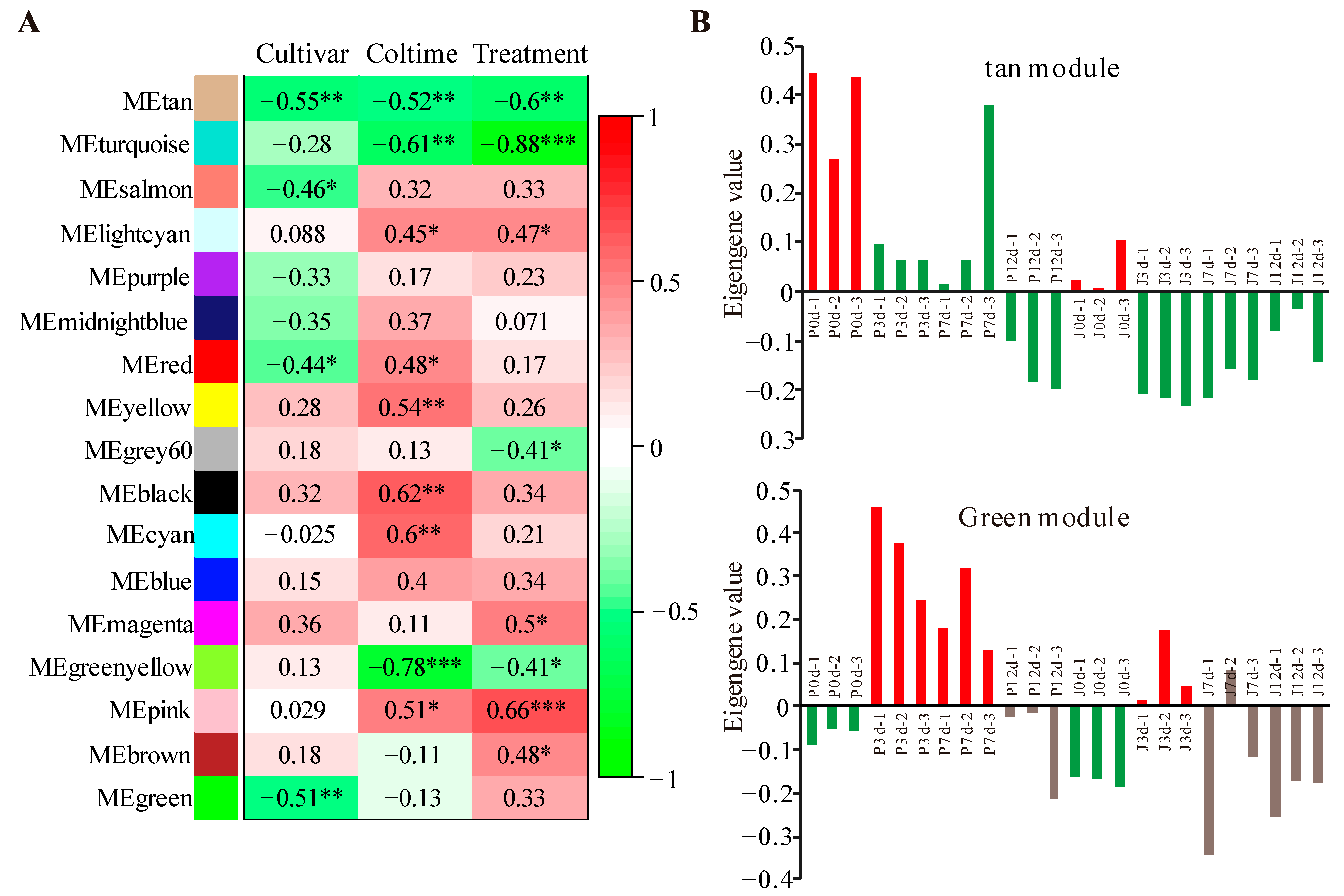
3.2. Modules Associated with Grafting Traits
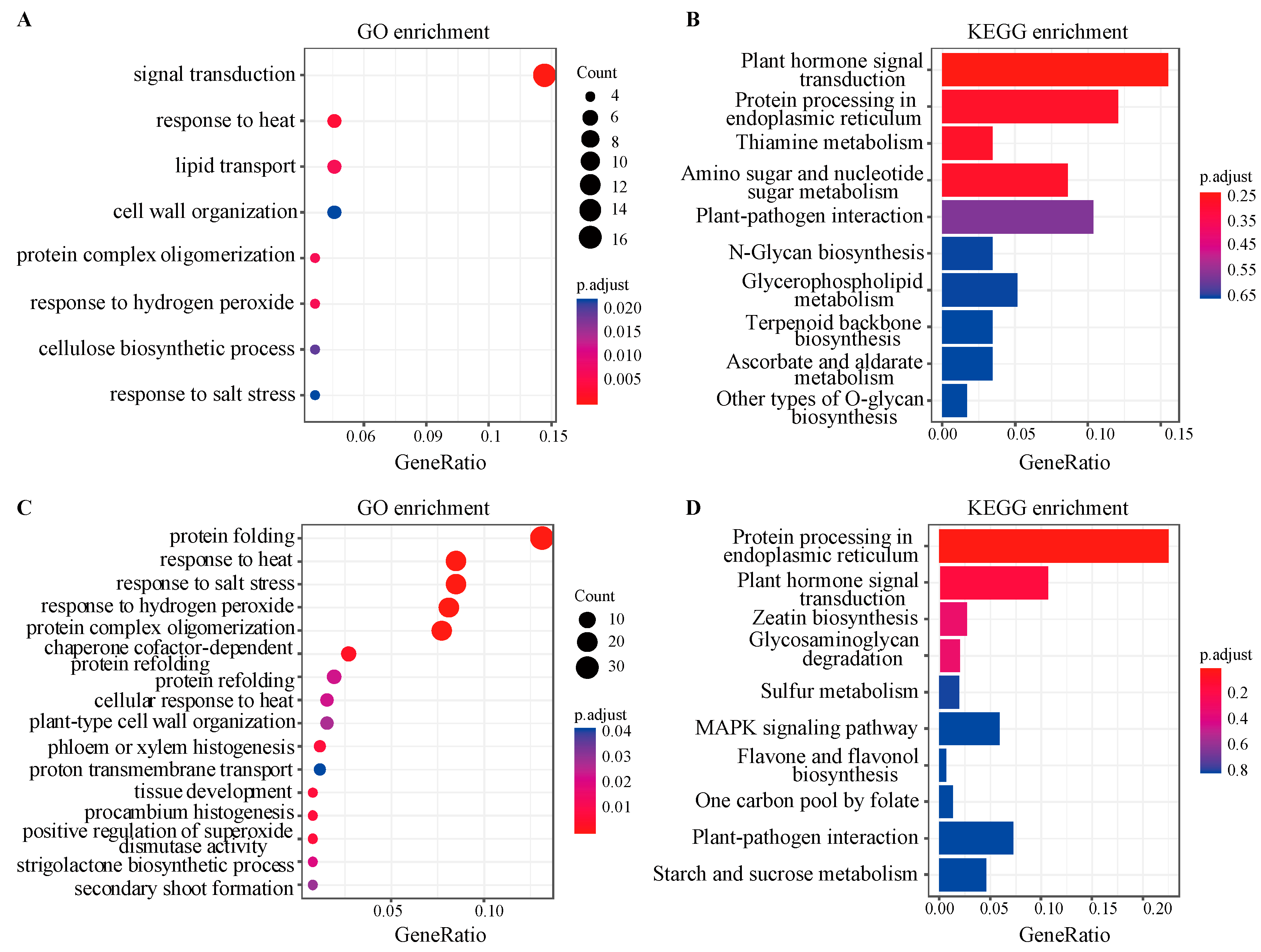
3.3. Functional Enrichment Analysis of Biologically Significant Module
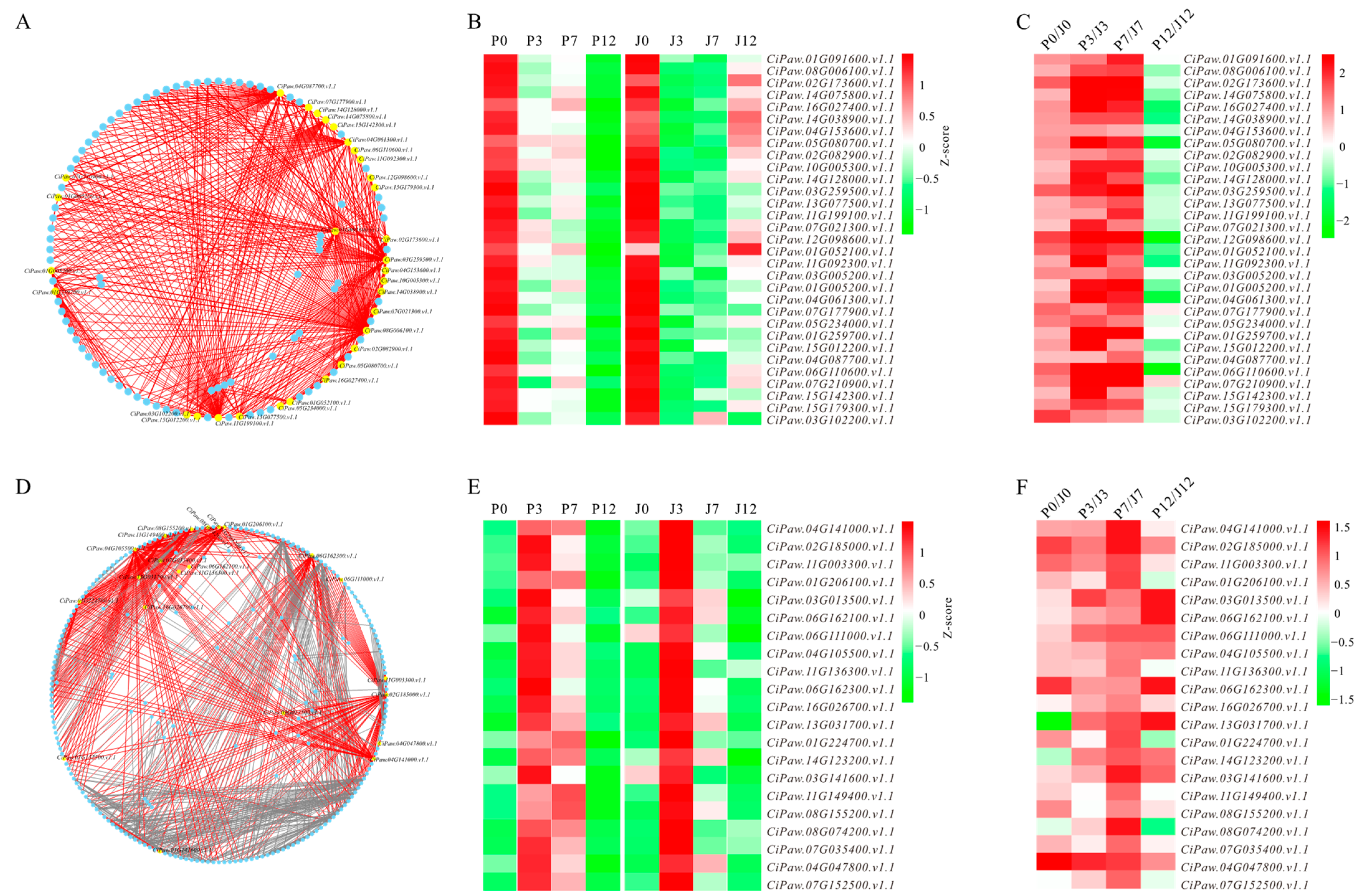
3.4. Identification of Hub Genes from Biologically Significant Modules
3.5. Promoter Variants of Hub Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Melnyk, C.W. Plant grafting: Insights into tissue regeneration. Regeneration 2017, 4, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Loupit, G.; Cookson, S.J. Identifying molecular markers of successful graft union formation and compatibility. Front. Plant Sci. 2020, 11, 610352. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, P.; Binczycki, P.; Errea, P.; Martens, H.J.; Pina, A. Oxidative stress associated with rootstock–scion interactions in pear/quince combinations during early stages of graft development. J. Plant Physiol. 2015, 176, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Zhang, T.; Liu, J.; Sun, X.; Sun, X.; Wang, W.; Zheng, C. Interactions between rootstock and scion during grafting and their molecular regulation mechanism. Sci. Hortic. 2023, 308, 111554. [Google Scholar] [CrossRef]
- Mo, Z.; Feng, G.; Su, W.; Liu, Z.; Peng, F. Transcriptomic analysis provides insights into grafting union development in pecan (Carya illinoinensis). Genes 2018, 9, 71. [Google Scholar] [CrossRef]
- Yuan, H.; Zhao, L.; Qiu, L.; Xu, D.; Tong, Y.; Guo, W.; Yang, X.; Shen, C.; Yan, D.; Zheng, B. Transcriptome and hormonal analysis of grafting process by investigating the homeostasis of a series of metabolic pathways in Torreya grandis cv. Merrillii. Ind. Crop. Prod. 2017, 108, 814–823. [Google Scholar] [CrossRef]
- Assunção, M.; Santos, C.; Brazão, J.; Eiras-Dias, J.; Fevereiro, P. Understanding the molecular mechanisms underlying graft success in grapevine. BMC Plant Biol. 2019, 19, 396. [Google Scholar] [CrossRef]
- Liang, C.; Sayed Haidar Abbas, R.; Yu, S.; Zuhair, M.M.; Abdullah, F.S.; Fayez, M.S.; Muna, O.A.; Bandar, H.A.; Ahmed Mohajja, A.; Schreurs, N.M.; et al. Bioinformatics role of the WGCNA analysis and co-expression network identifies of prognostic marker in lung cancer. Saudi J. Biol. Sci. 2022, 29, 3519–3527. [Google Scholar]
- Wu, J.; Zhao, X.; Lin, Z.; Shao, Z. A system level analysis of gastric cancer across tumor stages with RNA-seq data. Mol. Biosyst. 2015, 11, 1925–1932. [Google Scholar] [CrossRef]
- Friedman, N.; Linial, M.; Nachman, I.; Pe’er, D. Using Bayesian networks to analyze expression data. J. Comput. Biol. 2000, 7, 601–620. [Google Scholar] [CrossRef]
- Segal, E.; Shapira, M.; Regev, A.; Pe’er, D.; Botstein, D.; Koller, D.; Friedman, N. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003, 34, 166–176. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Pu, L.; Wang, M.; Li, K.; Feng, T.; Zheng, P.; Li, S.; Yao, Y.; Jin, L. Identification micro-RNAs functional modules and genes of ischemic stroke based on weighted gene co-expression network analysis (WGCNA). Genomics 2020, 112, 2748–2754. [Google Scholar] [CrossRef] [PubMed]
- Ai, D.; Wang, Y.; Li, X.; Pan, H. Colorectal cancer prediction based on weighted gene co-expression network analysis and variational auto-encoder. Biomolecules 2020, 10, 1207. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Y.; Zhang, Z.; Li, H. Identification of KIF18B as a hub candidate gene in the metastasis of clear cell renal cell carcinoma by weighted gene co-expression network analysis. Front. Genet. 2020, 11, 905. [Google Scholar] [CrossRef]
- Yao, Q.; Song, Z.; Wang, B.; Qin, Q.; Zhang, J. Identifying key genes and functionally enriched pathways in Sjögren’s syndrome by weighted gene co-expression network analysis. Front. Genet. 2019, 10, 1142. [Google Scholar] [CrossRef]
- Li, Z.X.; Zhang, W.L.; Jue, D.W.; Liu, X.; Jiang, Y.S.; Tang, J.M. Transcriptome changes induced by Botrytis cinerea stress and weighted gene co-expression network analysis (WGCNA) in Actinidia chinensis. Plant Mol. Biol. Rep. 2022, 40, 389–401. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, X.; Zhou, J.; Deng, H.; Zhang, G.; Xiao, Y.; Tang, W. WGCNA analysis identifies the hub genes related to heat stress in seedling of rice (Oryza sativa L.). Genes 2022, 13, 1020. [Google Scholar] [CrossRef]
- Xie, L.; Dong, C.; Shang, Q. Gene co-expression network analysis reveals pathways associated with graft healing by asymmetric profiling in tomato. BMC Plant Biol. 2019, 19, 373. [Google Scholar] [CrossRef]
- Fan, Y.; Zheng, Y.; Teixeira da Silva, J.A.; Yu, X. Comparative transcriptomics and WGCNA reveal candidate genes involved in petaloid stamens in Paeonia lactiflora. J. Hortic. Sci. Biotech. 2021, 96, 588–603. [Google Scholar] [CrossRef]
- Mo, Z.; Chen, Y.; Lou, W.; Jia, X.; Zhai, M.; Xuan, J.; Guo, Z.; Li, Y. Identification of suitable reference genes for normalization of real-time quantitative PCR data in pecan (Carya illinoinensis). Trees 2020, 34, 1233–1241. [Google Scholar] [CrossRef]
- Bentley, N.; Grauke, L.; Ruhlman, E.; Klein, R.R.; Kubenka, K.; Wang, X.; Klein, P. Linkage mapping and QTL analysis of pecan (Carya illinoinensis) full-siblings using genotyping-by-sequencing. Tree Genet. Genomes 2020, 16, 83. [Google Scholar] [CrossRef]
- Zhang, R.; Peng, F.; Li, Y. Pecan production in China. Sci. Hortic. 2015, 197, 719–727. [Google Scholar] [CrossRef]
- Lovell, J.T.; Bentley, N.B.; Bhattarai, G.; Jenkins, J.W.; Sreedasyam, A.; Alarcon, Y.; Bock, C.; Boston, L.B.; Carlson, J.; Cervantes, K. Four chromosome scale genomes and a pan-genome annotation to accelerate pecan tree breeding. Nat. Commun. 2021, 12, 4125. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Chen, J.; Qing, C.; He, S.; Zou, C.; Yuan, G.; Yang, C.; Peng, H.; Pan, G. GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor. Appl. Genet. 2021, 134, 3305–3318. [Google Scholar] [CrossRef]
- Ko, D.K.; Brandizzi, F. Network-based approaches for understanding gene regulation and function in plants. Plant J. 2020, 104, 302–317. [Google Scholar] [CrossRef]
- Muneer, S.; Ko, C.H.; Soundararajan, P.; Manivnnan, A.; Park, Y.G.; Jeong, B.R. Proteomic study related to vascular connections in watermelon scions grafted onto bottle-gourd rootstock under different light intensities. PLoS ONE 2015, 10, e0120899. [Google Scholar] [CrossRef]
- Mo, Z.; He, H.; Su, W.; Peng, F. Analysis of differentially accumulated proteins associated with graft union formation in pecan (Carya illinoensis). Sci. Hortic. 2017, 224, 126–134. [Google Scholar] [CrossRef]
- Rezaee, R.; Vahdati, K.; Grigoorian, V.; Valizadeh, M. Walnut grafting success and bleeding rate as affected by different grafting methods and seedling vigour. J. Hortic. Sci. Biotech. 2008, 83, 94–99. [Google Scholar] [CrossRef]
- Ahmad, P.; Sarwat, M.; Sharma, S. Reactive oxygen species, antioxidants and signaling in plants. J. Plant Biol. 2008, 51, 167–173. [Google Scholar] [CrossRef]
- Aloni, B.; Karni, L.; Deventurero, G.; Levin, Z.; Cohen, R.; Katzir, N.; Lotan-Pompan, M.; Edelstein, M.; Aktas, H.; Turhan, E. Physiological and biochemical changes at the rootstock-scion interface in graft combinations between Cucurbita rootstocks and a melon scion. J. Hortic. Sci. Biotech. 2008, 83, 777–783. [Google Scholar] [CrossRef]
- Cookson, S.J.; Clemente Moreno, M.J.; Hevin, C.; Nyamba Mendome, L.Z.; Delrot, S.; Trossat-Magnin, C.; Ollat, N. Graft union formation in grapevine induces transcriptional changes related to cell wall modification, wounding, hormone signalling, and secondary metabolism. J. Exp. Bot. 2013, 64, 2997–3008. [Google Scholar] [CrossRef] [PubMed]
- Planas-Riverola, A.; Gupta, A.; Betegón-Putze, I.; Bosch, N.; Ibañes, M.; Caño-Delgado, A.I. Brassinosteroid signaling in plant development and adaptation to stress. Development 2019, 146, dev151894. [Google Scholar] [CrossRef]
- Jin, M.; Liu, Y.; Shi, B.; Yuan, H. Exogenous IAA improves the seedling growth of Syringa villosa via regulating the endogenous hormones and enhancing the photosynthesis. Sci. Hortic. 2023, 308, 111585. [Google Scholar] [CrossRef]
- Sharma, A.; Zheng, B. Molecular responses during plant grafting and its regulation by auxins, cytokinins, and gibberellins. Biomolecules 2019, 9, 397. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, Y.; Zhao, M.; Liu, Y.; Xu, X.; Li, T. Transcriptomic analysis of melon/squash graft junction reveals molecular mechanisms potentially underlying the graft union development. PeerJ 2021, 9, e12569. [Google Scholar] [CrossRef]
- Peng, Y.; Yang, J.; Li, X.; Zhang, Y. Salicylic acid: Biosynthesis and signaling. Annu. Rev. Plant Biol. 2021, 72, 761–791. [Google Scholar] [CrossRef]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The crosstalks between jasmonic acid and other plant hormone signaling highlight the involvement of jasmonic acid as a core component in plant response to biotic and abiotic stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef]
- Jang, G.; Yoon, Y.; Choi, Y.D. Crosstalk with jasmonic acid integrates multiple responses in plant development. Int. J. Mol. Sci. 2020, 21, 305. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Lambolez, A.; Kojima, M.; Takebayashi, Y.; Heyman, J.; Watanabe, S.; Seo, M.; De Veylder, L. Wounding triggers callus formation via dynamic hormonal and transcriptional changes. Plant Physiol. 2017, 175, 1158–1174. [Google Scholar] [CrossRef]
- Chang, H.C.; Chu, C.P.; Lin, S.J.; Hsiao, C.K. Network hub-node prioritization of gene regulation with intra-network association. BMC Bioinform. 2020, 21, 101. [Google Scholar] [CrossRef]
- Goncalves, A.; Leigh-Brown, S.; Thybert, D.; Stefflova, K.; Turro, E.; Flicek, P.; Brazma, A.; Odom, D.T.; Marioni, J.C. Extensive compensatory cis-trans regulation in the evolution of mouse gene expression. Genome Res. 2012, 22, 2376–2384. [Google Scholar] [CrossRef] [PubMed]
- Mack, K.L.; Nachman, M.W. Gene regulation and speciation. Trends Genet. 2017, 33, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Ng, D.W.; Zhang, C.; Comai, L.; Ye, W.; Jeffrey Chen, Z. Cis-and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012, 3, 950. [Google Scholar] [CrossRef]
- Tripathi, P.; Rabara, R.C.; Rushton, P.J. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 2014, 239, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Tian, N.; She, F.; Cao, A.; Wu, W.; Zheng, S.; Yang, N. Characteristics analysis of early responsive to dehydration genes in Arabidopsis thaliana (AtERD). Plant Signal. Behav. 2022, 2105021. [Google Scholar] [CrossRef]
- Yokotani, N.; Ichikawa, T.; Kondou, Y.; Matsui, M.; Hirochika, H.; Iwabuchi, M.; Oda, K. Expression of rice heat stress transcription factor OsHsfA2e enhances tolerance to environmental stresses in transgenic Arabidopsis. Planta 2008, 227, 957–967. [Google Scholar] [CrossRef]
- Rehman, S.; Mahmood, T. Functional role of DREB and ERF transcription factors: Regulating stress-responsive network in plants. Acta Physiol. Plant 2015, 37, 178. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Li, Q.; Chen, X.; Li, X. Comparative transcriptome analysis to elucidate the enhanced thermotolerance of tea plants (Camellia sinensis) treated with exogenous calcium. Planta 2019, 249, 775–786. [Google Scholar] [CrossRef]
- Breen, S.; Williams, S.J.; Outram, M.; Kobe, B.; Solomon, P.S. Emerging insights into the functions of pathogenesis-related protein 1. Trends Plant Sci. 2017, 22, 871–879. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, Z.; Jiang, X.; Zhang, Y.; Zhai, M.; Hu, L.; Xuan, J. Weighted Gene Co-Expression Network Analysis Reveals Key Pathways and Hub Genes Associated with Successful Grafting in Pecan (Carya illinoinensis). Forests 2023, 14, 835. https://doi.org/10.3390/f14040835
Mo Z, Jiang X, Zhang Y, Zhai M, Hu L, Xuan J. Weighted Gene Co-Expression Network Analysis Reveals Key Pathways and Hub Genes Associated with Successful Grafting in Pecan (Carya illinoinensis). Forests. 2023; 14(4):835. https://doi.org/10.3390/f14040835
Chicago/Turabian StyleMo, Zhenghai, Xiaozhuang Jiang, Yan Zhang, Min Zhai, Longjiao Hu, and Jiping Xuan. 2023. "Weighted Gene Co-Expression Network Analysis Reveals Key Pathways and Hub Genes Associated with Successful Grafting in Pecan (Carya illinoinensis)" Forests 14, no. 4: 835. https://doi.org/10.3390/f14040835
APA StyleMo, Z., Jiang, X., Zhang, Y., Zhai, M., Hu, L., & Xuan, J. (2023). Weighted Gene Co-Expression Network Analysis Reveals Key Pathways and Hub Genes Associated with Successful Grafting in Pecan (Carya illinoinensis). Forests, 14(4), 835. https://doi.org/10.3390/f14040835