
Fungal Community Succession of Populus grandidentata (Bigtooth Aspen) during Wood Decomposition

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Field Sampling
2.3. Sample Processing
2.4. DNA Extraction and Amplicon Sequencing
2.5. Quantitative PCR
2.6. Chemical Analyses
2.7. Extracellular Enzyme Activity (EEA)
2.8. Sequence Analyses
2.9. Additional Statistical Analysis
3. Results
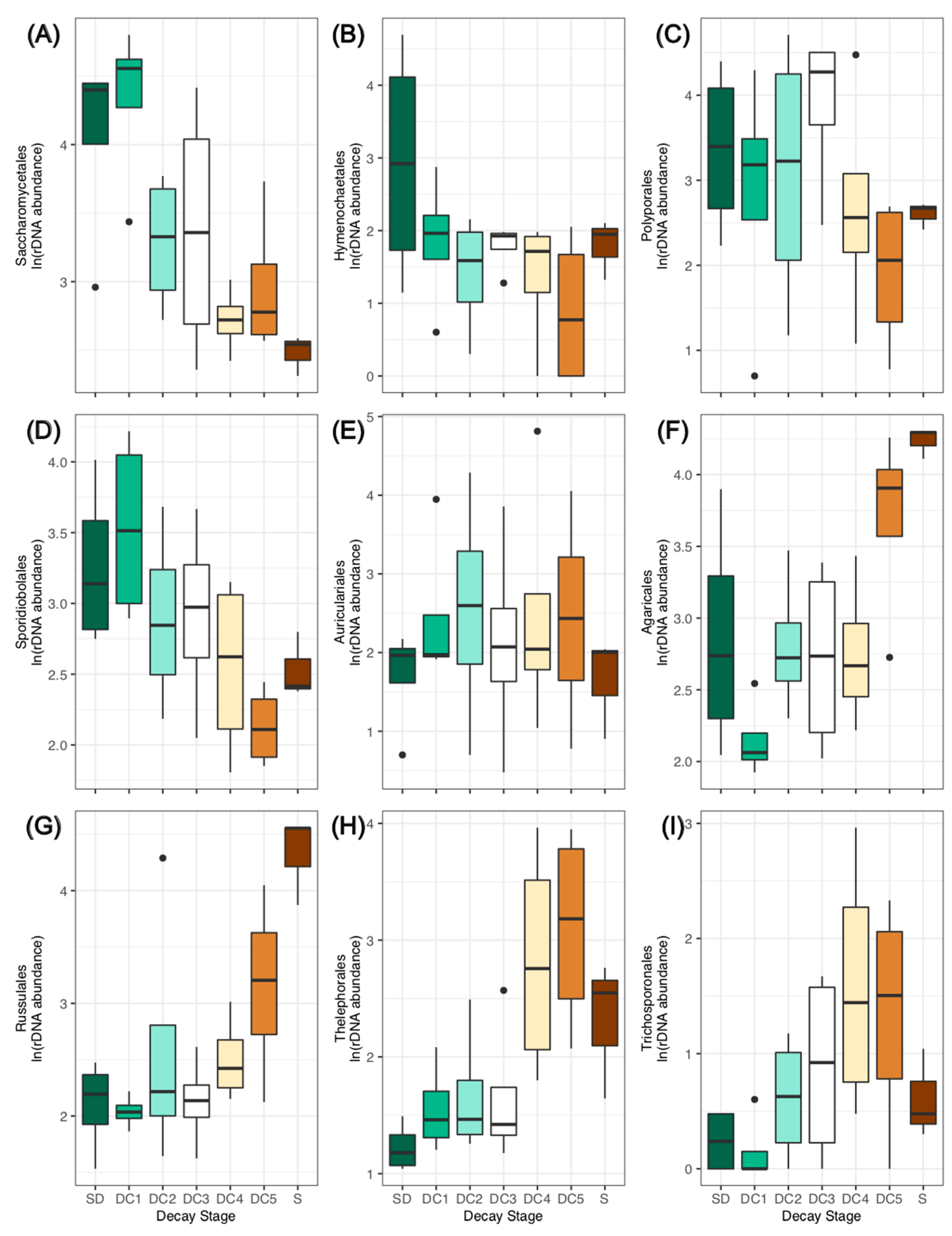
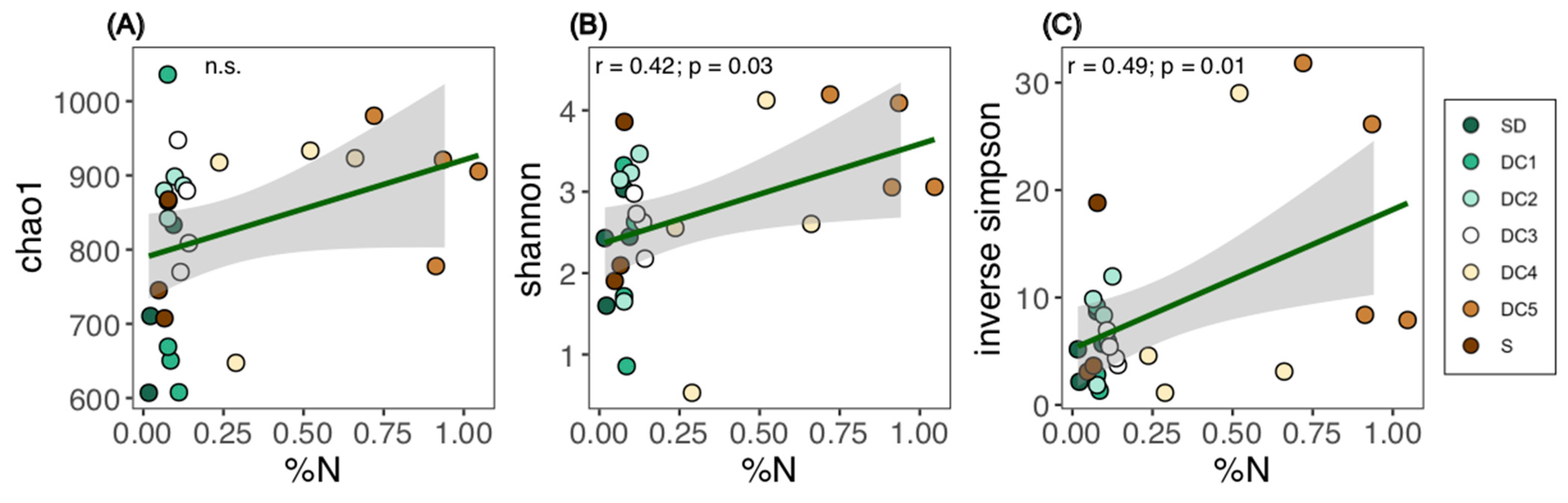
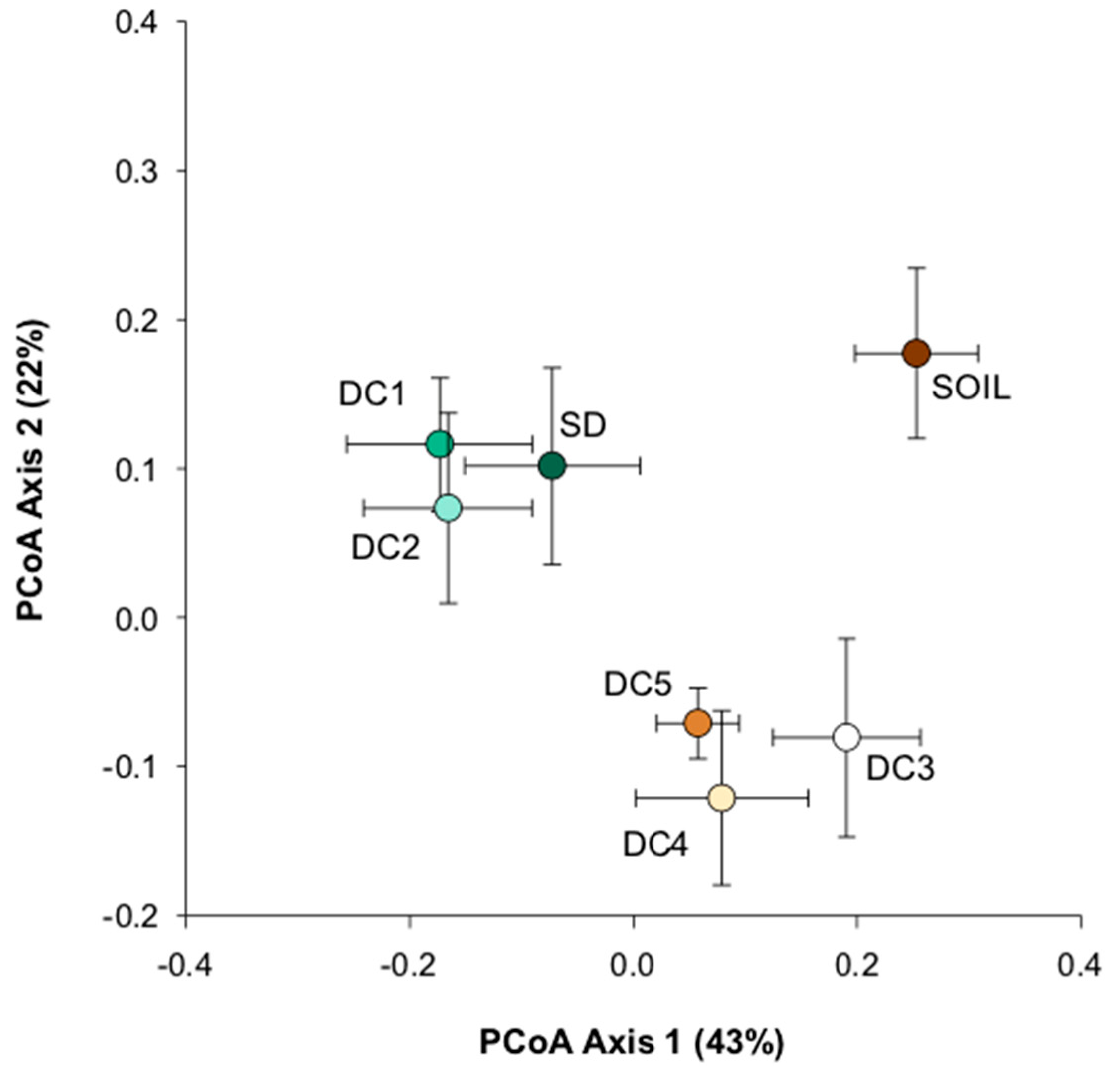
3.1. Fungal Community Structure
3.2. Wood Chemical Composition and Fungal Succession
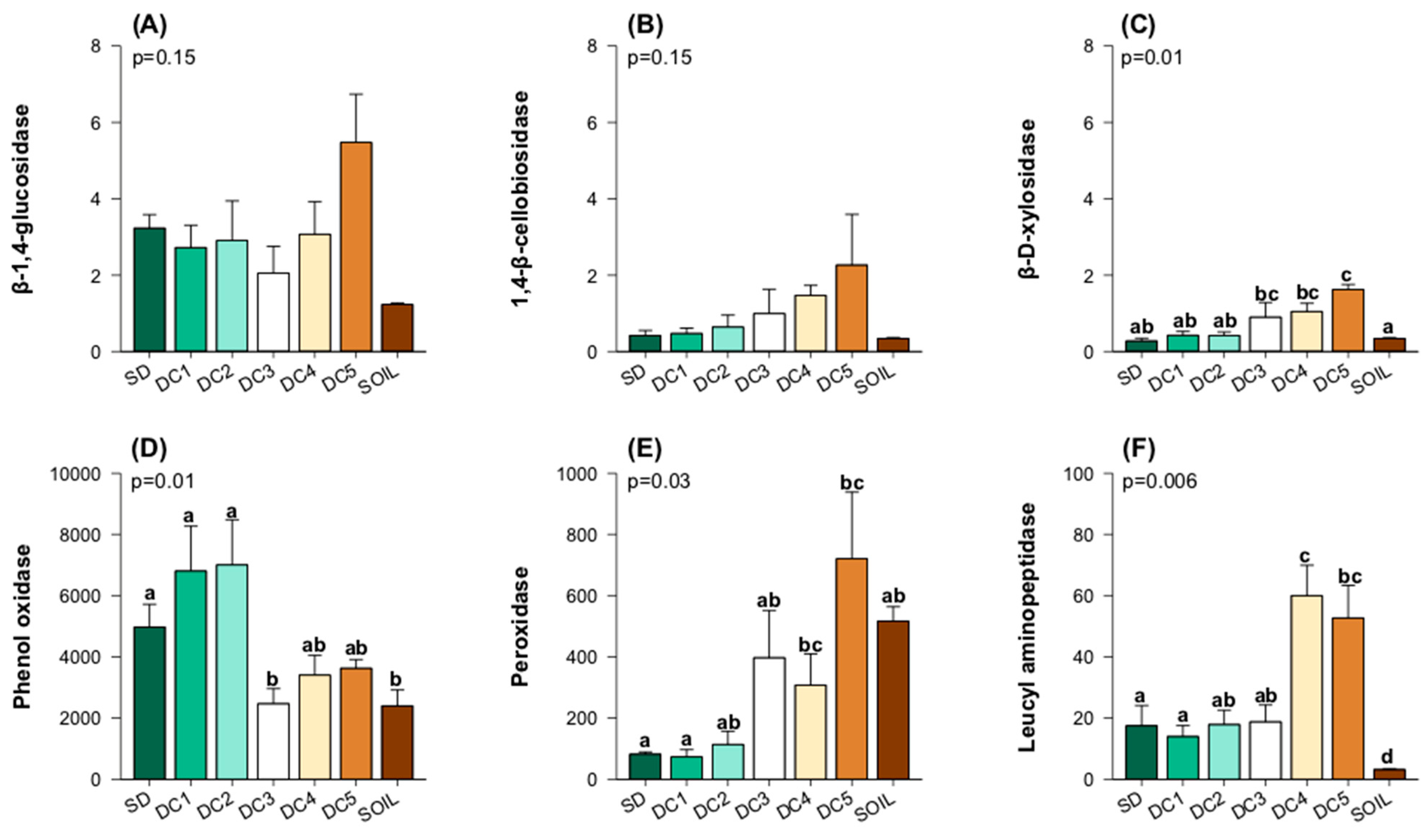
3.3. Enzyme Activity
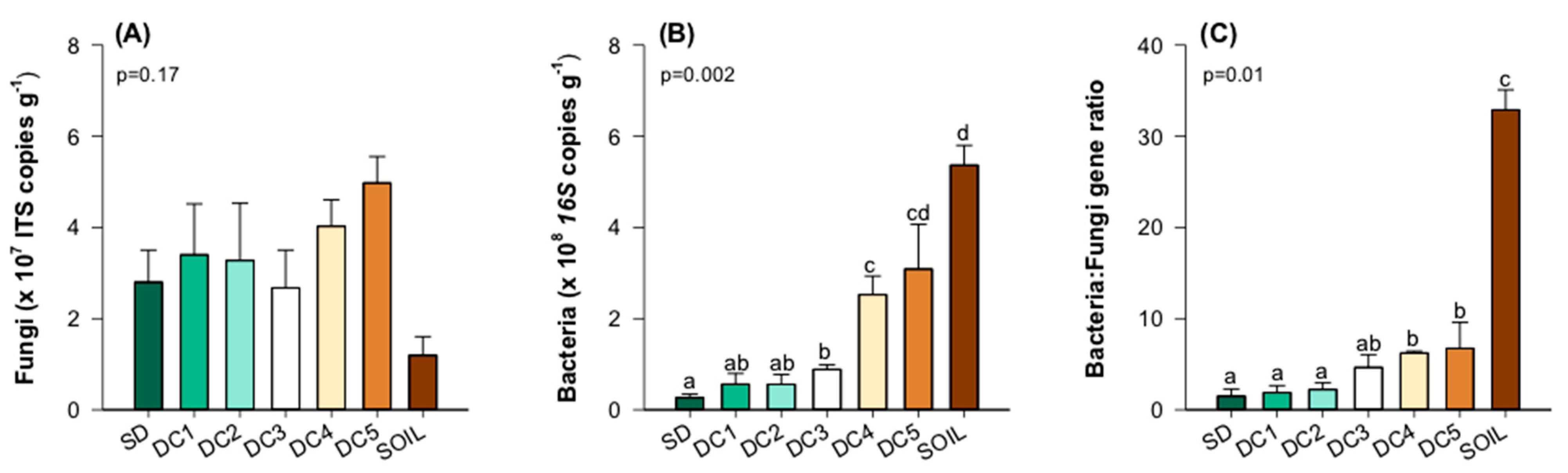
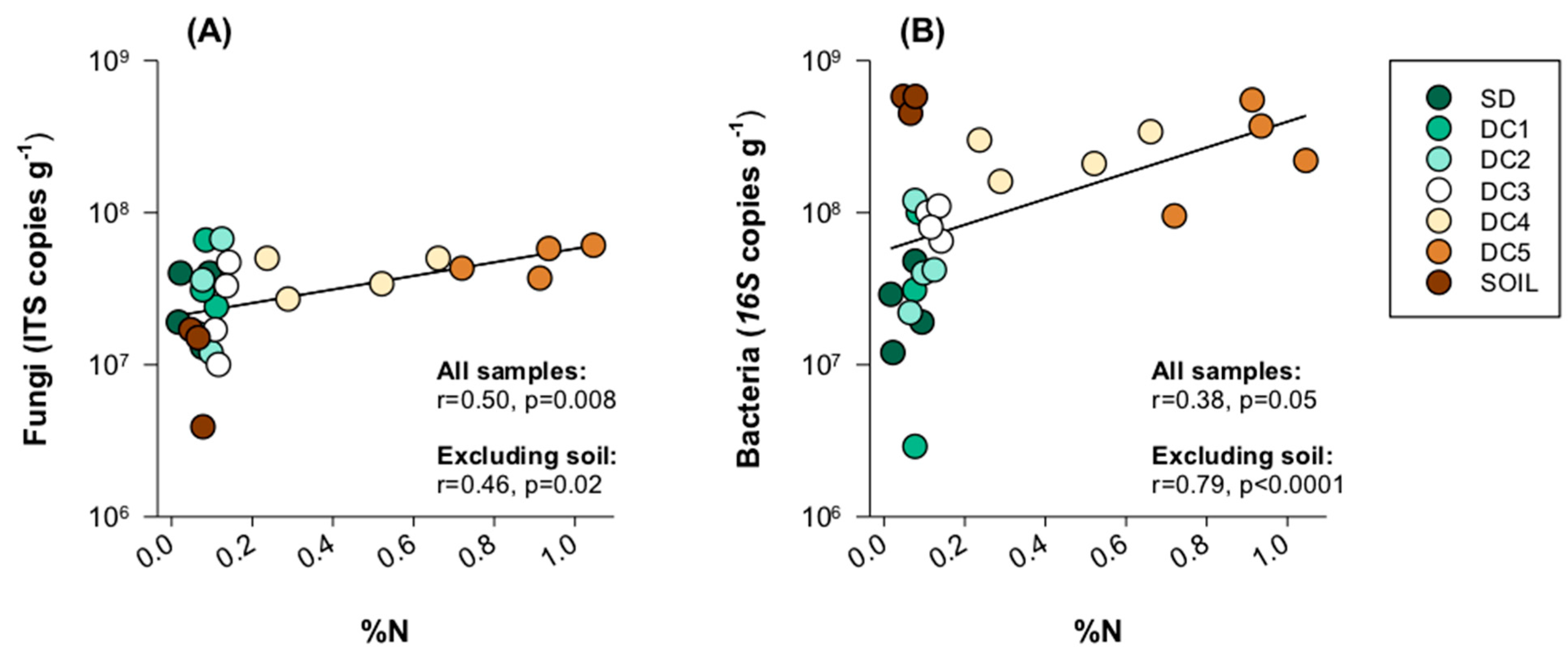
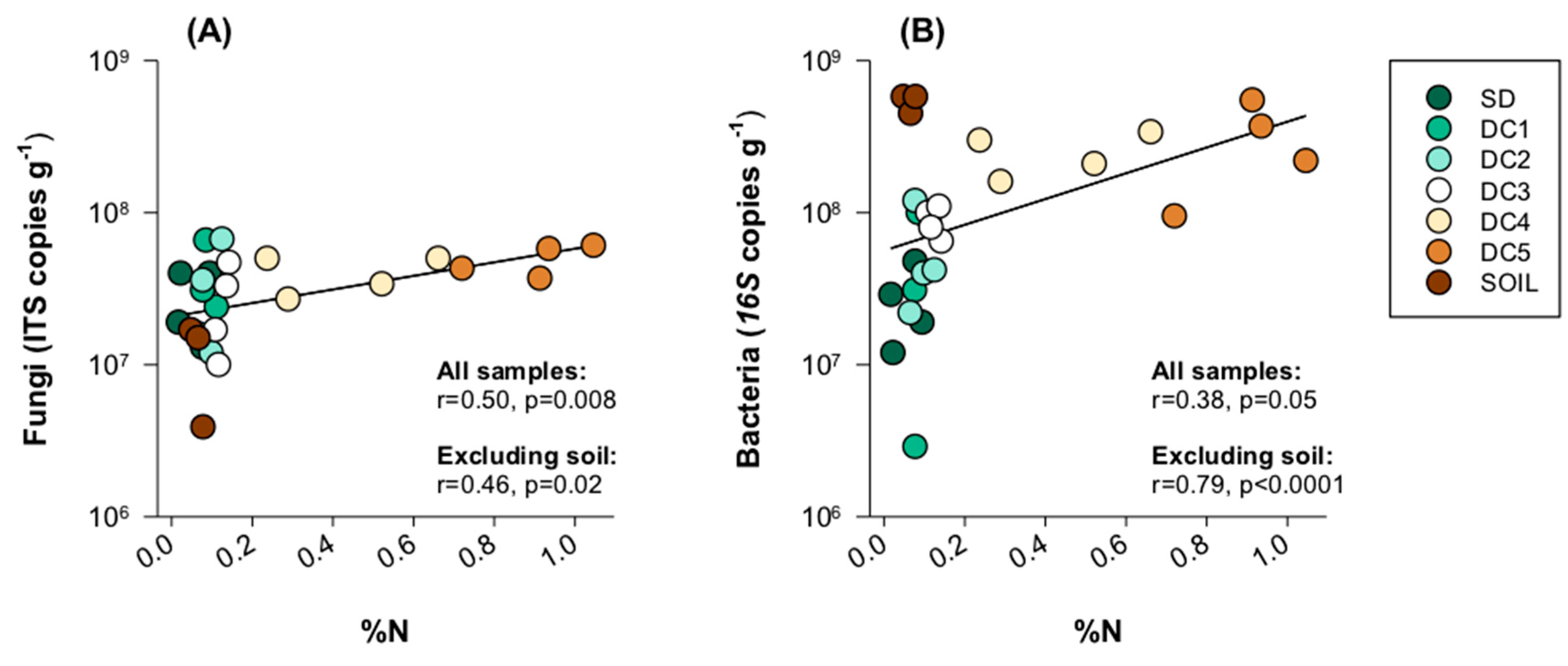
3.4. Fungal and Bacterial Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harmon, M.E.; Franklin, J.F.; Swanson, F.J.; Sollins, P.; Gregory, S.V.; Lattin, J.D.; Anderson, N.H.; Cline, S.P.; Aumen, N.G.; Sedell, J.R.; et al. Ecology of coarse woody debris in temperate ecosystems. Adv. Ecol. Res. 1986, 15, 133–302. [Google Scholar] [CrossRef]
- Forrester, D.I.; Pretzsch, H. Tamm Review: On the strength of evidence when comparing ecosystem functions of mixtures with monocultures. For. Ecol. Manag. 2015, 356, 41–53. [Google Scholar] [CrossRef]
- Brais, S.; Sadi, R.; Bergeron, Y.; Grenier, Y. Coarse woody debris dynamics in a post-fire jack pine chronosequence and its relation with site productivity. For. Ecol. Manag. 2005, 220, 216–226. [Google Scholar] [CrossRef]
- Gough, C.M.; Vogel, C.S.; Kazanski, C.; Nagel, L.; Flower, C.E.; Curtis, P.S. Coarse woody debris and the carbon balance of a north temperate forest. For. Ecol. Manag. 2007, 244, 60–67. [Google Scholar] [CrossRef]
- Forrester, J.A.; Mladenoff, D.J.; D’Amato, A.W.; Fraver, S.; Lindner, D.L.; Brazee, N.J.; Clayton, M.K.; Gower, S.T. Temporal trends and sources of variation in carbon flux from coarse woody debris in experimental forest canopy openings. Oecologia 2015, 179, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.V.; Vogel, C.S.; Liebman, E.; Curtis, P.S.; Gough, C.M. Coarse woody debris and the carbon balance of a moderately disturbed forest. For. Ecol. Manag. 2016, 361, 38–45. [Google Scholar] [CrossRef]
- Harmon, M.E.; Bond-Lamberty, B.; Tang, J.W.; Vargas, R. Heterotrophic respiration in disturbed forests: A review with examples from North America. J. Geophys. Res. Biogeosci. 2011, 116, G00k04. [Google Scholar] [CrossRef]
- Toljander, Y.K.; Lindahl, B.D.; Holmer, L.; Högberg, N.O. Environmental fluctuations facilitate species co-existence and increase decomposition in communities of wood decay fungi. Oecologia 2006, 148, 625–631. [Google Scholar] [CrossRef]
- Valentin, L.; Rajala, T.; Peltoniemi, M.; Heinonsalo, J.; Pennanen, T.; Mäkipää, R. Loss of diversity in wood-inhabiting fungal communities affects decomposition activity in Norway spruce wood. Front. Micro. 2014, 5, 230. [Google Scholar] [CrossRef]
- Kirk, T.K.; Cowling, E.B. Degradation by white rot fungi of phenolic model compounds related to lignin. Phytopathology 1968, 58, 1055. [Google Scholar]
- Moll, J.; Kellner, H.; Leonhardt, S.; Stengel, E.; Dahl, A.; Bässler, C.; Buscot, F.; Hofrichter, M.; Hoppe, B. Bacteria inhabiting deadwood of 13 tree species are heterogeneously distributed between sapwood and heartwood. Environ Microbiol. 2018, 20, 3744–3756. [Google Scholar] [CrossRef]
- Floudas, D. Evolution of lignin decomposition systems in fungi. Wood degradation and ligninolytic fungi. Adv. Bot. Res. 2021, 99, 37–76. [Google Scholar] [CrossRef]
- Van der Wal, A.; Geydan, T.D.; Kuyper, T.W.; de Boer, W. A thready affair: Linking fungal diversity and community dynamics to terrestrial decomposition processes. FEMS Microbiol. Rev. 2013, 37, 477–494. [Google Scholar] [CrossRef]
- Gonzalez-Perez, E.; Yanez-Morales, M.J.; Santiago-Santiago, V.; Montero-Pineda, A. Fungi biodiversity on pepper wilt and some related factors, in Tlacotepec de Jost Manzo, El Verde, Puebla. Agrociencia 2004, 38, 653–661. [Google Scholar]
- Bonnarme, P.; Jeffries, T.W. MN(II) Regulation of lignin peroxidases and manganese-dependent peroxidases from lignin-degrading white rot fungi. Appl. Environ. Microbiol. 1990, 56, 210–217. [Google Scholar] [CrossRef]
- Hatakka, A. Lignin-modifying enzymes from selected white-rot fungi—Production and role in lignin degradation. FEMS Microbiol. Rev. 1994, 13, 125–135. [Google Scholar] [CrossRef]
- Eisenlord, S.D.; Freedman, Z.; Zak, D.R.; Xue, K.; He, Z.L.; Zhou, J.Z. Microbial mechanisms mediating increased soil C storage under elevated atmospheric N deposition. Appl. Environ. Microbiol. 2013, 79, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Y.; Wang, Y.M.; Zhang, L.L.; Xu, M. Morphological and mechanical properties of tannic acid/PAAm semi-IPN hydrogels for cell adhesion. Polym. Test. 2017, 61, 314–323. [Google Scholar] [CrossRef]
- Kubartová, A.; Ottosson, E.; Dahlberg, A.; Stenlid, J. Patterns of fungal communities among and within decaying logs, revealed by 454 sequencing. Mol. Ecol. 2012, 21, 4514–4532. [Google Scholar] [CrossRef]
- Song, Z.W.; Kennedy, P.G.; Liew, F.J.; Schilling, J.S. Fungal endophytes as priority colonizers initiating wood decomposition. Funct. Ecol. 2017, 31, 407–418. [Google Scholar] [CrossRef]
- Fukami, T.; Dickie, I.A.; Wilkie, J.P.; Paulus, B.C.; Park, D.; Roberts, A.; Buchanan, P.K.; Allen, R.B. Assembly history dictates ecosystem functioning: Evidence from wood decomposer communities. Ecol. Lett. 2010, 13, 675–684. [Google Scholar] [CrossRef]
- Holmer, L.; Stenlid, J. Diffuse competition for heterogeneous substrate in soil among six species of wood-decomposing basidiomycetes. Oecologia 1996, 106, 531–538. [Google Scholar] [CrossRef]
- Jonsson, M.; Wardle, D.A. Context dependency of litter-mixing effects on decomposition and nutrient release across a long-term chronosequence. Oikos 2008, 117, 1674–1682. [Google Scholar] [CrossRef]
- Boddy, L.; Tordoff, G.M.; Wood, J.; Hynes, J.; Bebber, D.; Jones, T.H.; Fricker, M. Mycelial foraging strategies of saprotrophic cord-forming basidiomycetes. In Proceedings of the 8th International Mycological Congress of the International-Mycological-Association (IMC8), Cairns, Australia, 20–25 August 2006. [Google Scholar]
- Boddy, L.; Hynes, J.; Bebber, D.P.; Fricker, M.D. Saprotrophic cord systems: Dispersal mechanisms in space and time. Mycoscience 2009, 50, 9–19. [Google Scholar] [CrossRef]
- Hamzeh, M.; Dayanandan, S. Phylogeny of Populus (Salicaceae) based on nucleotide sequences of chloroplast TRNT-TRNF region and nuclear rDNA. Am. J. Bot. 2004, 91, 1398–1408. [Google Scholar] [CrossRef]
- Baldrian, P.; Zrůstová, P.; Tláskal, V.; Davidová, A.; Merhautová, V.; Vrška, T. Fungi associated with decomposing deadwood in a natural beech-dominated forest. Fungal Ecol. 2016, 23, 109–122. [Google Scholar] [CrossRef]
- Mäkipää, R.; Rajala, T.; Schigel, D.; Rinne, K.T.; Pennanen, T.; Abrego, N.; Ovaskainen, O. Interactions between soil-and dead wood-inhabiting fungal communities during the decay of Norway spruce logs. ISME J. 2017, 11, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Rajala, T.; Tuomivirta, T.; Pennanen, T.; Mäkipää, R. Habitat models of wood-inhabiting fungi along a decay gradient of Norway spruce logs. Fungal Ecol. 2015, 18, 48–55. [Google Scholar] [CrossRef]
- Hoppe, B.; Kahl, T.; Karasch, P.; Wubet, T.; Bauhus, J.; Buscot, F.; Krüger, D. Network analysis reveals ecological links between N-fixing bacteria and wood-decaying fungi. PLoS ONE 2014, 9, e88141. [Google Scholar] [CrossRef]
- Sun, H.; Terhonen, E.; Kasanen, R.; Asiegbu, F.O. Diversity and community structure of primary wood-inhabiting bacteria in boreal forest. Geomicrobiol. J. 2014, 31, 315–324. [Google Scholar] [CrossRef]
- Arnstadt, T.; Hoppe, B.; Kahl, T.; Kellner, H.; Krüger, D.; Bässler, C.; Bauhus, J.; Hofrichter, M. Patterns of laccase and peroxidases in coarse woody debris of Fagus sylvatica, Picea abies and Pinus sylvestris and their relation to different wood parameters. Eur. J. For. Res. 2016, 135, 109–124. [Google Scholar] [CrossRef]
- Kuramae, E.E.; Leite, M.F.A.; Suleiman, A.K.A.; Gough, C.M.; Castillo, B.T.; Faller, L.; Franklin, R.B.; Syring, J. Wood decay characteristics and interspecific interactions control bacterial community succession in Populus grandidentata (Bigtooth Aspen). Front. Microbiol. 2019, 10, 979. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.S.; Zhao, W.Q.; Xiao, J.; Zhang, Z.L.; Qiao, M.F.; Liu, Q.; Yin, H. Exudate components exert different influences on microbially mediated C losses in simulated rhizosphere soils of a spruce plantation. Plant Soil. 2017, 419, 127–140. [Google Scholar] [CrossRef]
- Gough, C.M.; Vogel, C.S.; Hardiman, B.; Curtis, P.S. Wood net primary production resilience in an unmanaged forest transitioning from early to middle succession. For. Ecol. Manag. 2010, 260, 36–41. [Google Scholar] [CrossRef]
- Lapin, M.; Barnes, B.V. Using the landscape ecosystem approach to assess species and ecosystem diversity. Conserv. Biol. 1995, 9, 1148–1158. [Google Scholar] [CrossRef]
- Nave, L.E.; Sparks, J.P.; Le Moine, J.; Hardiman, B.S.; Nadelhoffer, K.J.; Tallant, J.M.; Vogel, C.S.; Strahm, B.D.; Curtis, P.S. Changes in soil nitrogen cycling in a northern temperate forest ecosystem during succession. Biogeochemistry 2014, 121, 471–488. [Google Scholar] [CrossRef]
- Harmon, M.E.; Fasth, B.G.; Yatskov, M.; Kastendick, D.; Rock, J.; Woodall, C.W. Release of coarse woody detritus-related carbon: A synthesis across forest biomes. Carbon Balance Manag. 2020, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, E.M.; Berrier, D.J.; Neubauer, S.C.; Franklin, R.B. Using microbial communities and extracellular enzymes to link soil organic matter characteristics to greenhouse gas production in a tidal freshwater wetland. Biogeochemistry 2014, 117, 473–490. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Orsello, C.E.; Lauffenburger, D.A.; Hammer, D.A. Molecular properties in cell adhesion: A physical and engineering perspective. Trends Biotechnol. 2001, 19, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef]
- Ottosson, E.; Kubartova, A.; Edman, M.; Jönsson, M.; Lindhe, A.; Stenlid, J.; Dahlberg, A. Diverse ecological roles within fungal communities in decomposing logs of Picea abies. FEMS Microbiol. Ecol. 2015, 91, fiv012. [Google Scholar] [CrossRef] [PubMed]
- Kubartova, A.; Ottosson, E.; Stenlid, J. Linking fungal communities to wood density loss after 12 years of log decay. FEMS Microbiol. Ecol. 2015, 91, fiv032. [Google Scholar] [CrossRef]
- Van der Wal, A.; Ottosson, E.; de Boer, W. Neglected role of fungal community composition in explaining variation in wood decay rates. Ecology 2015, 96, 124–133. [Google Scholar] [CrossRef]
- Brischke, C.; Rapp, A.O. Influence of wood moisture content and wood temperature on fungal decay in the field: Observations in different micro-climates. Wood Sci. Technol. 2008, 42, 663–677. [Google Scholar] [CrossRef]
- Blanchette, R.A. Delignification by wood-decaying fungi. Annu. Rev. Phytopathol. 1991, 29, 381–398. [Google Scholar] [CrossRef]
- Bowman, D.; Balch, J.K.; Artaxo, P.; Bond, W.J.; Carlson, J.M.; Cochrane, M.A.; D’Antonio, C.M.; DeFries, R.S.; Doyle, J.C.; Harrison, S.P.; et al. Fire in the earth system. Science 2009, 324, 481–484. [Google Scholar] [CrossRef]
- Johnston, S.R.; Boddy, L.; Weightman, A.J. Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol. Ecol. 2016, 92, fiw179. [Google Scholar] [CrossRef]
- TlÁskal, V.; Brabcová, V.; Větrovský, T.; Jomura, M.; López-Mondéjar, R.; Monteiro, L.M.O.; Saraiva, J.P.; Human, Z.R.; Cajthaml, T.; Nunes da Rocha, U.; et al. Complementary roles of wood-inhabiting fungi and bacteria facilitate deadwood decomposition. Msystems 2021, 6, 10–1128. [Google Scholar] [CrossRef]
- Suh, S.O.; Blackwell, M.; Kurtzman, C.P.; Lachance, M.A. Phylogenetics of Saccharomycetales, the ascomycete yeasts. Mycologia 2006, 98, 1006–1017. [Google Scholar] [CrossRef]
- Sampaio, J.P.; Gonçalves, P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl. Environ. Microbiol. 2008, 74, 2144–2152. [Google Scholar] [CrossRef]
- Bhadra, B.; Rao, R.S.; Singh, P.K.; Sarkar, P.K.; Shivaji, S. Yeasts and yeast-like fungi associated with tree bark: Diversity and identification of yeasts producing extracellular endoxylanases. Curr. Microbiol. 2008, 56, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Kurtzman, C.P.; Fell, J.W.; Boekhout, T. Definition, classification and nomenclature of the yeasts. In Yeasts: A Taxonomic Study, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 1–3, pp. 3–5. [Google Scholar] [CrossRef]
- Johnson, M.; East, D.A.; Mulvihill, D.P. Formins determine the functional properties of actin filaments in yeast. Curr. Biol. 2014, 24, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Viner, I.; Spirin, V.; Zíbarová, L.; Larsson, K.H. Additions to the taxonomy of Lagarobasidium and Xylodon (Hymenochaetales, Basidiomycota). Mycokeys 2018, 41, 65–90. [Google Scholar] [CrossRef]
- Blanchette, R.A.; Shaw, C.G. Associations among bacteria, yeasts, and basidiomycetes during wood decay. Phytopath 1978, 68, 631–637. [Google Scholar] [CrossRef]
- Fricker, M.D.; Bebber, D.; Boddy, L. Mycelial networks: Structure and dynamics. In British Mycological Society Symposia; Ecol of Saprotrophic Basidiomycetes; Academic Press: Cambridge, MA, USA, 2008; Volume 28, pp. 3–18. [Google Scholar]
- Mašínová, T.; Bahnmann, B.D.; Větrovský, T.; Tomšovský, M.; Merunková, K.; Baldrian, P. Drivers of yeast community composition in the litter and soil of a temperate forest. FEMS Microbiol. Ecol. 2017, 93, fiw223. [Google Scholar] [CrossRef] [PubMed]
- Middelhoven, W.J.; Scorzetti, G.; Fell, J.W. Trichosporon porosum comb. nov., an anamorphic basidiomycetous yeast inhabiting soil, related to the loubieri/laibachii group of species that assimilate hemicelluloses and phenolic compounds. FEMS Yeast Res. 2001, 1, 15–22. [Google Scholar]
- Yurkov, A.; Krüger, D.; Begerow, D.; Arnold, N.; Tarkka, M.T. Basidiomycetous yeasts fromboletales fruiting bodies and their interactions with the mycoparasite Sepedonium chrysospermum and the Host Fungus Paxillus. Microb. Ecol. 2012, 63, 295–303. [Google Scholar] [CrossRef]
- Averill, C. Slowed decomposition in ectomycorrhizal ecosystems is independent of plant chemistry. Soil Biol. Biochem. 2016, 102, 52–54. [Google Scholar] [CrossRef]
- Zak, D.R.; Pellitier, P.T.; Argiroff, W.A.; Castillo, B.; James, T.Y.; Nave, L.E.; Averill, C.; Beidler, K.V.; Bhatnagar, J.; Blesh, J.; et al. Exploring the role of ectomycorrhizal fungi in soil carbon dynamics. New Phytol. 2019, 223, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pellitier, P.T.; Zak, D.R. Ectomycorrhizal fungi and the enzymatic liberation of nitrogen from soil organic matter: Why evolutionary history matters. New Phytol. 2018, 217, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, H.; Denef, K.; Six, J.; Frey, S.D.; Merckx, R.; Paustian, K. Influence of microbial populations and residue quality on aggregate stability. Appl. Soil Ecol. 2001, 16, 195–208. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Six, J.; Frey, S.D.; Thiet, R.K.; Batten, K.M. Bacterial and fungal contributions to carbon sequestration in agroecosystems. Soil Sci. Soc. Am. J. 2006, 70, 555–569. [Google Scholar] [CrossRef]
- Hoppe, B.; Krüger, D.; Kahl, T.; Arnstadt, T.; Buscat, F.; Bauhus, J.; Wubet, T. A pyrosequencing insight into sprawling bacterial diversity and community dynamics in decaying deadwood logs of Fagus sylvatica and Picea abies. Sci. Rep. 2015, 5, 9456. [Google Scholar] [CrossRef]
- Rousk, J.; Brookes, P.C.; Bååth, E. Investigating the mechanisms for the opposing pH relationships of fungal and bacterial growth in soil. Soil Biol. Biochem. 2010, 42, 926–934. [Google Scholar] [CrossRef]
- Nacke, H.; Goldmann, K.; Schöning, I.; Pfeiffer, B.; Kaiser, K.; Castillo-Villamizar, G.A.; Schrumpf, M.; Buscot, F.; Daniel, R.; Wubet, T. Fine spatial scale variation of soil microbial communities under european beech and Norway spruce. Front. Microbiol. 2016, 7, 2067. [Google Scholar] [CrossRef]
- Kahl, T.; Arnstadt, T.; Baber, K.; Bässler, C.; Bauhus, J.; Borken, W.; Buscot, F.; Floren, A.; Heibl, C.; Hessenmöller, D.; et al. Wood decay rates of 13 temperate tree species in relation to wood properties, enzyme activities and organismic diversities. 2017. For. Ecol. Manag. 2017, 391, 86–95. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SD | DC1 | DC2 | DC3 | DC4 | DC5 | |
|---|---|---|---|---|---|---|
| Soil | 0.03 ** | 0.02 ** | 0.02 ** | 0.02 ** | 0.03 ** | 0.03 ** |
| SD | - | 0.75 | 0.12 | 0.03 ** | 0.04 ** | 0.03 ** |
| DC1 | - | - | 0.58 | 0.32 | 0.06 * | 0.08 * |
| DC2 | - | - | - | 0.77 | 0.61 | 0.62 |
| DC3 | - | - | - | - | 0.75 | 0.22 |
| DC4 | - | - | - | - | - | 0.88 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo, B.T.; Franklin, R.B.; Amses, K.R.; Leite, M.F.A.; Kuramae, E.E.; Gough, C.M.; James, T.Y.; Faller, L.; Syring, J. Fungal Community Succession of Populus grandidentata (Bigtooth Aspen) during Wood Decomposition. Forests 2023, 14, 2086. https://doi.org/10.3390/f14102086
Castillo BT, Franklin RB, Amses KR, Leite MFA, Kuramae EE, Gough CM, James TY, Faller L, Syring J. Fungal Community Succession of Populus grandidentata (Bigtooth Aspen) during Wood Decomposition. Forests. 2023; 14(10):2086. https://doi.org/10.3390/f14102086
Chicago/Turabian StyleCastillo, Buck T., Rima B. Franklin, Kevin R. Amses, Márcio F. A. Leite, Eiko E. Kuramae, Christopher M. Gough, Timothy Y. James, Lewis Faller, and John Syring. 2023. "Fungal Community Succession of Populus grandidentata (Bigtooth Aspen) during Wood Decomposition" Forests 14, no. 10: 2086. https://doi.org/10.3390/f14102086
APA StyleCastillo, B. T., Franklin, R. B., Amses, K. R., Leite, M. F. A., Kuramae, E. E., Gough, C. M., James, T. Y., Faller, L., & Syring, J. (2023). Fungal Community Succession of Populus grandidentata (Bigtooth Aspen) during Wood Decomposition. Forests, 14(10), 2086. https://doi.org/10.3390/f14102086