

Genetic Evaluation in Natural Populations of the Threatened Conifer Amentotaxus argotaenia (Hance) Pilg. (Taxaceae) Using Microsatellites
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
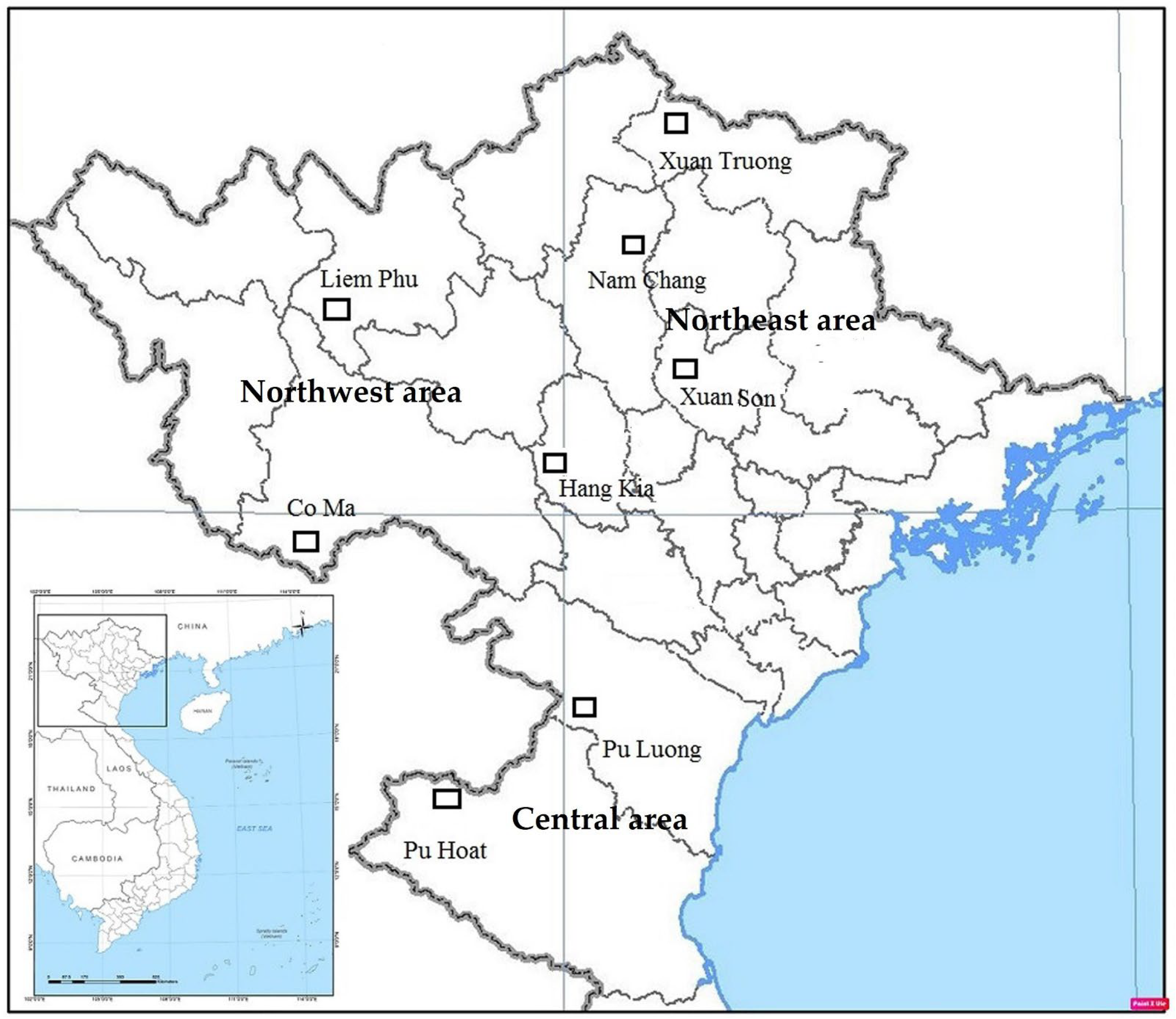
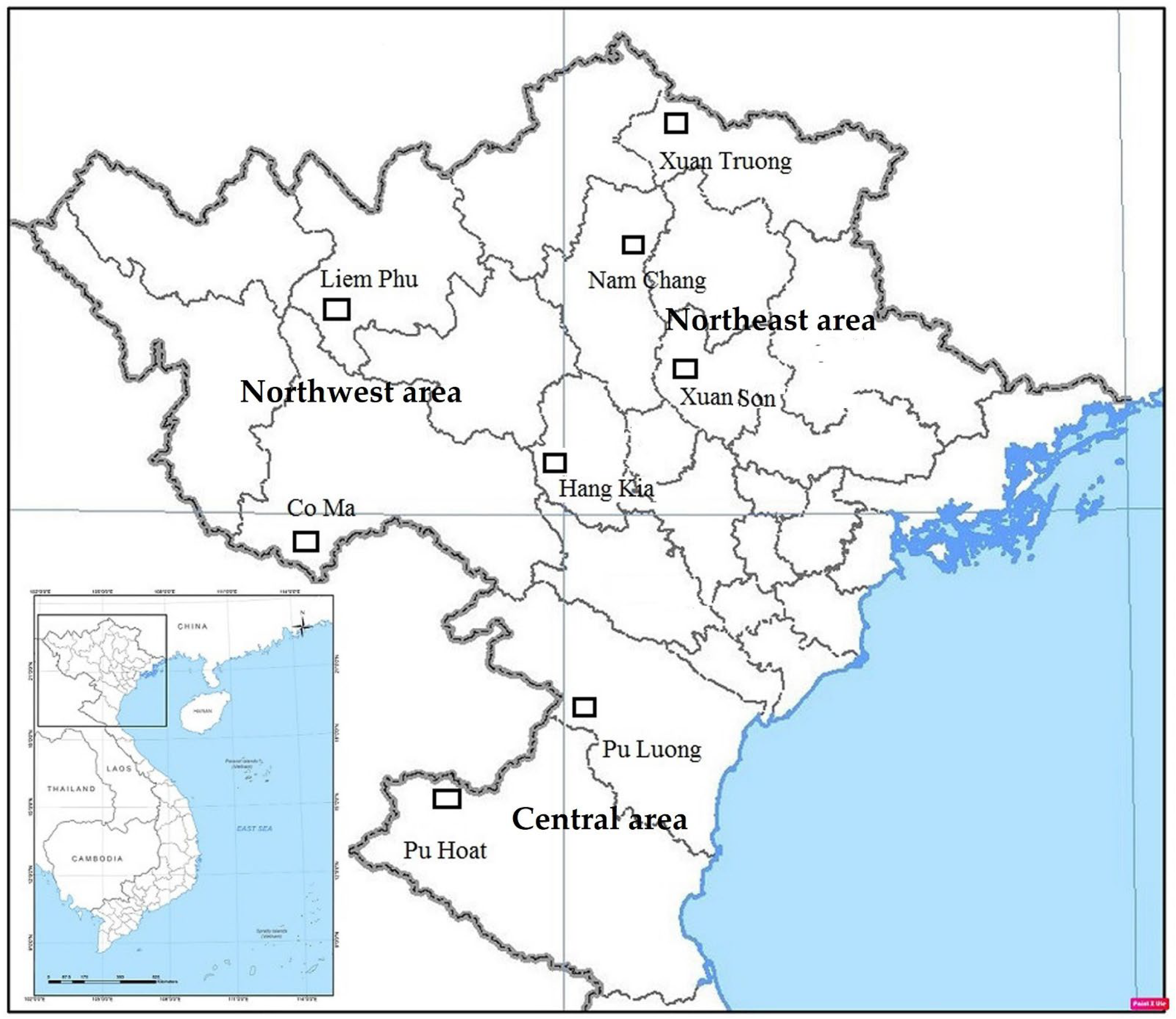
2.1. Study Sites and Tree Sample Collection
2.2. DNA Extraction and Microsatellite Amplification
2.3. Molecular Analysis, Genetic Diversity and Population Genetic Structure
3. Results
3.1. Genetic Diversity
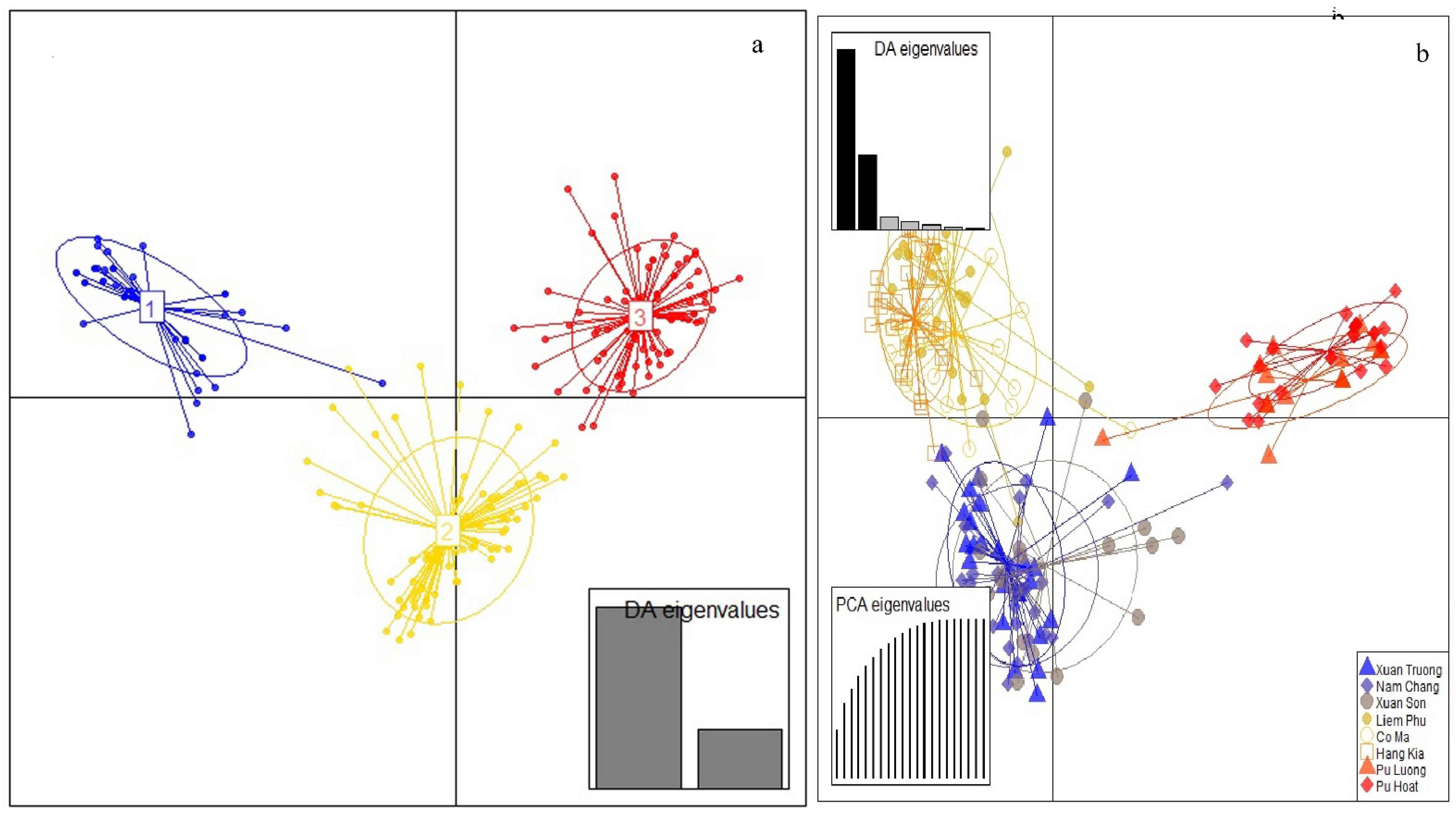
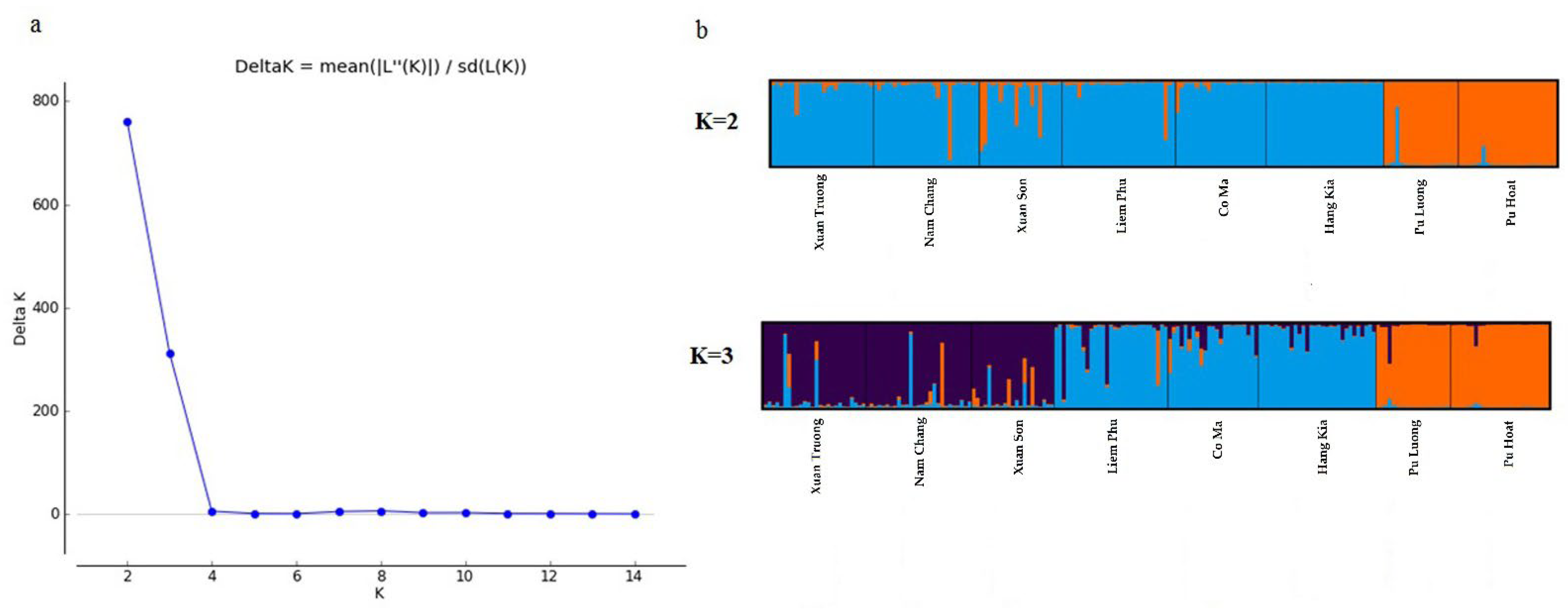
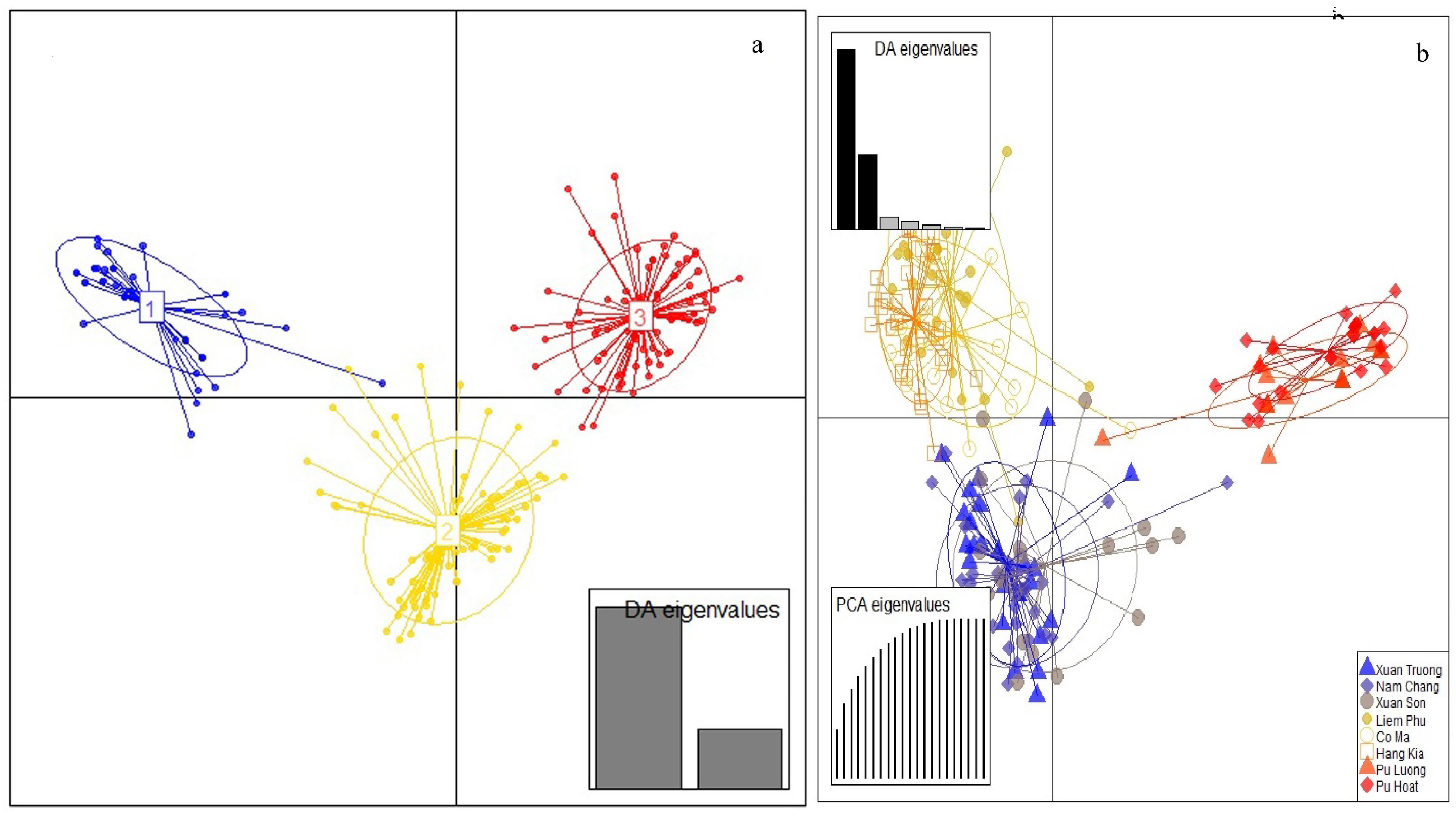
3.2. Genetic Structure
4. Discussion
4.1. Genetic Diversity
4.2. Population Genetic Structure
4.3. Conservation of Amentotaxus argotaenia in Vietnam
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hiep, N.T.; Loc, P.K.; Luu, N.D.T.; Thomas, P.L.; Farjon, A.; Averyanov, L.; Regalado, J., Jr. Vietnam Conifers: Conservation Status Review 2004; Science and Technics Publishing House: Hanoi, Vietnam, 2004. (In Vietnamese) [Google Scholar]
- Huey-Jen, S.; Lin, C.N.; Day, S.H.; Wang, L.W. A diterpenoid with a new skeleton and cytotoxic terpenoids isolated from Amentotaxus formosama. Helv. Chim. Acta 2003, 86, 2645–2652. [Google Scholar] [CrossRef]
- Hilton-Taylor, C.; Yang, Y.; Rushforth, K.; Liao, W. Amentotaxus argotaenia. IUCN Red List. Threat. Species 2013, e.T42545A2986540. [Google Scholar] [CrossRef]
- [BKHCN] Ministry of Science and Technology (Vietnam). Red Book of Vietnam, Part II Plants; Science and Technics Publishing House: Hanoi, Vietnam, 2007. (In Vietnamese)
- Charles, E.F.; Jeremie, B.F.; Hoban, S.; Kathleen, S.K.; Steger, L.; Aubihl, E.; Miquel, A.G.; Forry, S.; Hille, A.; Alejandro, A.R. Optimizing conservation strategies for a threatened tree species: In situ conservation of white ash (Fraxinus americana L.) genetic diversity through insecticide treatment. Forests 2018, 9, 202. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors influencing levels of genetic diversity in woody plant species. New For. 1992, 6, 95–124. [Google Scholar] [CrossRef]
- Frankham, R.; Bailou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2004; Volume 190, pp. 385–386. [Google Scholar]
- Lira, C.F.; Cardoso, S.R.; Ferreira, P.C.; Cardoso, M.A.; Provan, J. Long-term population isolation in the endangered tropical tree species Caesalpinia echinata Lam. revealed by chloroplast microsatellites. Mol. Ecol. 2003, 12, 3219–3225. [Google Scholar] [CrossRef]
- Bijlsma, P.; van Arendonk, J.A.M.; Woolliam, J.A. A general procedure for predict rates of inbreeding in populations undergoing mass selection. Genetics 2000, 154, 1865–1877. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Effects of history traits on genetic diversity in plant species. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 1996, 351, 1683–1685. [Google Scholar]
- Mihai, G.; Teodosiu, M. Genetic diversity and breeding of latch (Larix decidua Mill.) in Romania. Ann. For. Res. 2009, 52, 97–108. [Google Scholar]
- Bruford, M.W.; Davies, N.; Dulloo, M.E.; Faith, D.P.; Walter, M. Monitoring changes in genetic diversity. In The GEO Handbook on Biodiversity Observation Networks; Walter, M., Scholes, R.J., Eds.; Springer International Publishing: New York, NY, USA, 2017; pp. 107–128. [Google Scholar]
- Vu, D.D.; Bui, T.T.X.; Nguyen, M.T.; Vu, D.G.; Nguyen, M.D.; Bui, V.T.; Huang, X.; Zhang, Y. Genetic variation in two threatened species: North taxad pine (Taxus chinensis) and south taxad pine (Taxus wallichiana). J. For. Res. 2017, 28, 265–272. [Google Scholar] [CrossRef]
- Hou, L.; Cui, Y.; Li, X.C.W.; Zhang, Z.; Pang, X.; Li, Y. Genetic evaluation of natural populations of the endangered conifer Thuja koraiensis using microsatellite markers by restriction-associsted DNA sequencing. Genes 2018, 9, 218. [Google Scholar] [CrossRef]
- Ju, T.; Farhat, P.; Tao, W.; Miao, J.L.J.; Fan, X.; Tso, S.; Mao, K. Development and chareacterization of EST-SSR markers for Juniperus squamata (Cupressaceae), an ecologically important conifer in Asian mountains. Silvae Genet. 2020, 69, 116–122. [Google Scholar] [CrossRef]
- Cho, Y.G.; Ishii, T.; Temnykh, S.; Chen, X.; Lipovich, L.; McCouch, S.R.; Park, W.D.; Ayres, N.; Cartinhour, S. Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 2000, 100, 713–722. [Google Scholar] [CrossRef]
- Hu, J.; Wang, L.; Li, J. Comparison of genomic SSR and EST-SSR markers for estimating genetic diversity in cucumber. Biol. Plant 2011, 55, 577–580. [Google Scholar] [CrossRef]
- Munoz-Falcon, J.E.; Vilanova, S.; Plazas, M.; Prohens, J. Diversity, relationships, and genetic fingerprinting of the Listada de Gandia eggplant landrace using genomic SSRs and EST-SSRs. Sci. Hortic. 2011, 129, 238–246. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Tam, N.M.; Duy, V.D.; Xuan, B.T.T.; Duc, N.M. Genetic variation and population structure in Chinese water pine (Glyptostrobus pensilis): A threatened species. Indian J. Biotechnol. 2013, 12, 499–503. [Google Scholar]
- Ge, X.J.; Zhou, X.L.; Li, Z.C.; Hsu, T.W.; Schaal, B.A.; Chiang, T.Y. Low genetic diversity and significant population structuring in the relict Amentotaxus argotaenia complex (Taxaceae) based on ISSR fingerprinting. J. Plant Res. 2005, 118, 415–422. [Google Scholar] [CrossRef]
- Tam, M.N.; Trang, N.T.P.; Hoa, N.T. Genetic diversity of an endangered species Fokienia hodginsii (Cupressaceae). Afr. J. Biotechnol. 2011, 10, 15838–15844. [Google Scholar] [CrossRef]
- Li, C.Y.; Chiang, T.Y.; Chiang, Y.C.; Hsu, H.M.; Ge, X.J.; Huang, C.C.; Chen, C.T.; Hung, K.H. Cross-species, amplifiable EST-SSR markers for Amentotaxus species obtained by next-generation sequencing. Molecules 2016, 21, 67. [Google Scholar] [CrossRef]
- Stojnic, S.; Avramidou, E.V.; Fussi, B.; Westergren, M.; Orlovic, S.; Matovic, B.; Trudic, B.; Kraigher, H.; Aravanopoulos, F.A.; Konnert, M. Assessment of genetic diversity and population genetic structure of Norway spruce (Picea abies (L.) Karsten) at its southern lineage in Europe. Implications for conservation of forest genetic resources. Forests 2019, 10, 258. [Google Scholar] [CrossRef]
- Ge, X.J.; Hung, K.H.; Ko, Y.Z.; Hsu, T.W.; Gong, X.; Chiang, T.Y.; Chiang, Y.C. Genetic divergence and biogeographical patterns in Amentotaxus argotaenia species complex. Plant Mol. Biol. Rep. 2015, 33, 264–280. [Google Scholar] [CrossRef]
- Ho, C.S.; Chen, C.T.; Ko, Y.Z.; Ge, X.J.; Hung, K.H.; Hsu, T.W.; Chiang, Y.C. Isolation and characterization of 15 microsatellite loci in four endangered Amentotaxus species (Taxaceae). Am. J. Bot. 2012, 99, e157–e159. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, L.J. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, D.F. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 435–538. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3). 2001. Available online: http://www.unil.ch/izea/softwares/fstat.html (accessed on 1 August 2022).
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficient. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornnet, J.M. Bottleneck: A computer program for detecting recent reductions in the effective population size frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Weir, B.; Cookerham, C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Hedrick, P.W. A standardized genetic differentiation measure. Evolution 2005, 59, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Excoffer, L.; Laval, G.; Schneider, S. Arlequin v. 3.5. an integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar]
- Takezaki, N.; Nei, M.; Tamura, K. Software for constructing population trees from allele frequency data and computing other population statistics with windows interface. Mol. Evol. 2010, 27, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von-Holdt, B.M. Structure Harvester: A website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Res. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Puechmaille, S.J. The program structure does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Res. 2016, 16, 608–627. [Google Scholar] [CrossRef]
- Li, Y.L.; Liu, J.X. StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Res. 2018, 18, 176–177. [Google Scholar] [CrossRef]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A. CLUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Hamrick, J.L. The distribution of genetic variation within and among natural plant populations. In Genetics and Conservation; Schenwald-Cox, C.M., Chamber, S.M., MacBryde, B., Thomas, L., Eds.; The Benjamin/Cummings Publishing Company, Inc.: Menio Park, CA, USA, 1983; pp. 335–348. [Google Scholar]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Lang, X.; Li, S.; Su, J.; Wang, Y. Characterization of 15 polymorphic microsatellite loci for Cephalotaxus oliveri (Cephalotaxaceae), a conifer of medicinal importance. Int. J. Mol. Sci. 2012, 13, 11165–11172. [Google Scholar] [CrossRef] [PubMed]
- Gajurel, J.P.; Cornejo, C.; Werth, S.; Shrestha, K.K.; Scheidedegger, C. Development and characterization of microsatellite loci in the endangered species Taxus wallichiana (Taxaceae). Appl. Plant Sci. 2013, 1, 1200281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Q.; Zhou, N. Genetic diversity and population structure of the endangered conifer Taxus wallichiana var. mairei (Taxaceae) revealed by simple sequence repeat (SSR) markers. Biochem. Syst. Ecol. 2013, 49, 107–114. [Google Scholar] [CrossRef]
- Gramazio, P.; Plesa, J.; Truta, A.M.; Sestras, A.; Vilanova, S.; Plazas, M.; Vicente, O.; Boscaiu, M.; Prohens, J.; Sestra, R.E. Highly onformation SSR genotyping reveals large genetic diversity and limited differentiation in European larch (Larix decidua) populations from Romania. Turk. J. Agric. For. 2018, 42, 165–175. [Google Scholar] [CrossRef]
- Dias, A.; Giovannelli, G.; Fady, B.; Spanu, I.; Vendramin, G.G.; Bagnoli, F.; Carvalho, A.; Silva, M.E.; Brito, J.L.; Lousada, J.L.; et al. Portuguese Pinus nigra J.F. Arnold populations: Genetic diversity, structure and relationships inferred by SSR markers. Ann. For. Sci. 2020, 77, 64. [Google Scholar] [CrossRef]
- Ding, M.; Meng, K.; Fan, Q.; Tan, W.; Liao, W.; Chen, S. Development and valadation of EST-SSR markers for Fokienia hodginsii (Cupressaceae). Appl. Plant Sci. 2017, 5, 1600152. [Google Scholar] [CrossRef]
- Castelan, P.M.; Cortes-Cruz, M.; Mendoza-Castillo, M.C.; Cruz-Izquierdo, S.; Lopez-Upton, J.; Padilla, I.S. Diversity and geneitc structure inferred with microsatellites in natural populations of Pseudotsuga menziesii (Mirb.) Franco (Pinaceae) in the Central region of Mexico. Forests 2019, 10, 101. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Su, Y.; Wang, T. EST-SSR-based landscape genetics of Pseudotaxus chienii, a tertiary relict conifer endemic to China. Ecol. Evol. 2021, 11, 9498–9515. [Google Scholar] [CrossRef]
- Tam, M.N.; Loc, P.K.; Duy, V.D. Genetic diversity in Xuan nha pine (Pinus armandii subsp. xuannhaensis L.K. Phan). Res. J. Biotechnol. 2015, 10, 30–36. [Google Scholar]
- Honnay, O.; Jacquemyn, H. Susceptibility of common and rare plant species to the genetic consequence of habitat fragmentation. Conserv. Biol. 2007, 21, 823–831. [Google Scholar] [CrossRef]
- Gijbels, P.; Hert, K.D.; Jacquemyn, H.; Honnay, O. Reduced fecundity and genetic diversity in small populations of rewarding versus deceptive orchd species: A meta-analysis. Ecol. Evol. 2015, 148, 153–159. [Google Scholar] [CrossRef]
- Falk, D.A.; Holsinger, K.E. (Eds.) Genetic and Conservation of Rare Plants; Oxford University Press: New York, NY, USA, 1991; p. 283. [Google Scholar]
- Slatkin, M.; Barton, N.H. A comparison of three indirect methods for estimating average levels of gene flow. Evolution 1989, 43, 1349–1368. [Google Scholar] [CrossRef]
- Brutting, C.; Wesche, K.; Mayer, S.; Hensen, I. Genetic diversity of six arable plants in relation to their Red List status. Biodiv. Conserv. 2012, 21, 745–761. [Google Scholar] [CrossRef]
- Petit, R.J.; Mousadik, E.I.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 1998, 12, 844–855. [Google Scholar] [CrossRef]
- Markert, J.A.; Chamolin, D.M.; Gutjahr-Gobell, R.; Grear, J.S.; Kuhn, A.; McGreevy, T.J.; Roth, A.; Bagley, M.J.; Nacci, D.E. Population genetic diversity and fitness in multiple environments. BMC Evol. Biol. 2010, 10, 205. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Loci | NA | AR | NE | HO | HE | FIS | PIC | Null Allele | FIT | FST | G’ST | Nm | PHWE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| amen1 | 4 | 2.7 | 1.32 | 0.150 | 0.222 | 0.221 | 0.391 | no | 0.694 | 0.546 | 0.715 | 0.208 | *** |
| amen2 | 4 | 2.7 | 1.93 | 0.419 | 0.464 | 0.234 | 0.449 | 0.109 | 0.228 | 0.145 | 0.258 | 1.475 | ** |
| amen3 | 3 | 2.4 | 1.83 | 0.425 | 0.444 | 0.078 | 0.387 | no | 0.126 | 0.088 | 0.137 | 2.587 | ns |
| amen4 | 3 | 2.9 | 2.05 | 0.427 | 0.448 | 0.119 | 0.466 | 0.091 | 0.188 | 0.148 | 0.257 | 1.444 | * |
| amen5 | 2 | 1.9 | 1.16 | 0.124 | 0.112 | −0.174 | 0.260 | no | 0.630 | 0.665 | 0.756 | 0.126 | *** |
| amen6 | 2 | 2 | 1.41 | 0.276 | 0.277 | −0.043 | 0.366 | no | 0.413 | 0.413 | 0.581 | 0.356 | *** |
| amen7 | 4 | 3.8 | 1.96 | 0.365 | 0.444 | 0.207 | 0.629 | 0.233 | 0.476 | 0.363 | 0.681 | 0.439 | *** |
| amen8 | 4 | 3 | 2.20 | 0.521 | 0.529 | 0.101 | 0.550 | 0.087 | 0.172 | 0.158 | 0.334 | 1.328 | *** |
| amen9 | 4 | 2.3 | 1.46 | 0.274 | 0.284 | 0.021 | 0.275 | no | 0.121 | 0.089 | 0.104 | 2.566 | ns |
| Mean | 1.70 | 0.331 (0.023) | 0.358 (0.022) | 0.120 | 0.419 | 0.339 (0.074) | 0.290 (0.071) | 0.443 | 1.170 (0.320) |
| Population | P% | NA | AR | AE | NP | HO (SE) | HE (SE) | FIS (SE) | FIS IIM | p Value of Bottleneck | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SMM | TPM | ||||||||||
| Xuan Truong | 100 | 2.6 | 2.4 | 1.8 | - | 0.329 | 0.386 | 0.166 ** | 0.092 | 0.248 | 0.125 |
| Nam Chang | 100 | 2.4 | 2.3 | 1.7 | - | 0.296 | 0.359 | 0.193 ** | 0.138 | 0.410 | 0.150 |
| Xuan Son | 100 | 2.6 | 2.4 | 1.8 | - | 0.354 | 0.421 | 0.181 ** | 0.077 | 0.213 | 0.064 |
| Liem Phu | 88.89 | 2.9 | 2.3 | 1.7 | 3 | 0.314 | 0.337 | 0.086 | 0.070 | 0.726 | 0.679 |
| Co Ma | 100 | 2.7 | 2.4 | 1.7 | - | 0.353 | 0.365 | 0.056 | 0.051 | 0.820 | 0.589 |
| Hang Kia | 88.89 | 2.3 | 2.1 | 1.6 | - | 0.341 | 0.313 | −0.083 | 0.022 | 0.527 | 0.320 |
| Pu Hoat | 100 | 2.4 | 2.4 | 1.8 | 1 | 0.347 | 0.382 | 0.074 | 0.093 | 0.875 | 0.715 |
| Pu Luong | 100 | 2.6 | 2.5 | 1.5 | - | 0.316 | 0.303 | −0.017 | 0.045 | 0.410 | 0.248 |
| Mean | 97.22 | 2.6 | 1.7 | 0.331 (0.023) | 0.358 (0.022) | 0.083 | 0.074 | ||||
| Df | Sum of Squares | Variance Components | Total Variation (%) | Fixation Indices | |
|---|---|---|---|---|---|
| Among areas | 2 | 221.904 | 0.820 | 32.49 | FST = 0.325 *** |
| Among populations within areas Within populations | 5 392 | 24.408 642.948 | 0.065 1.640 | 2.57 64.95 | |
| Total | 399 | 889.260 | 2.525 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.T.; La, T.T.; Do, X.T.; Do, H.V.; Nguyen, D.M.; Nguyen, H.P.L.; Murthy, H.N.; Phan, L.K.; Nguyen, T.M. Genetic Evaluation in Natural Populations of the Threatened Conifer Amentotaxus argotaenia (Hance) Pilg. (Taxaceae) Using Microsatellites. Forests 2022, 13, 1452. https://doi.org/10.3390/f13091452
Nguyen TT, La TT, Do XT, Do HV, Nguyen DM, Nguyen HPL, Murthy HN, Phan LK, Nguyen TM. Genetic Evaluation in Natural Populations of the Threatened Conifer Amentotaxus argotaenia (Hance) Pilg. (Taxaceae) Using Microsatellites. Forests. 2022; 13(9):1452. https://doi.org/10.3390/f13091452
Chicago/Turabian StyleNguyen, Thanh Trung, Thuy Thi La, Xuyen Thi Do, Hai Van Do, Duc Minh Nguyen, Hong Phan Lan Nguyen, Hosakatte Niranjana Murthy, Long Ke Phan, and Tam Minh Nguyen. 2022. "Genetic Evaluation in Natural Populations of the Threatened Conifer Amentotaxus argotaenia (Hance) Pilg. (Taxaceae) Using Microsatellites" Forests 13, no. 9: 1452. https://doi.org/10.3390/f13091452
APA StyleNguyen, T. T., La, T. T., Do, X. T., Do, H. V., Nguyen, D. M., Nguyen, H. P. L., Murthy, H. N., Phan, L. K., & Nguyen, T. M. (2022). Genetic Evaluation in Natural Populations of the Threatened Conifer Amentotaxus argotaenia (Hance) Pilg. (Taxaceae) Using Microsatellites. Forests, 13(9), 1452. https://doi.org/10.3390/f13091452