
Microsatellite Analysis Revealing High Genetic Diversity of the Chestnut Blight Fungus in South Tyrol (Northern Italy)


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Isolation of C. parasitica
2.2. DNA Extraction and Polymerase Chain Reaction (PCR)
2.3. Data Analysis
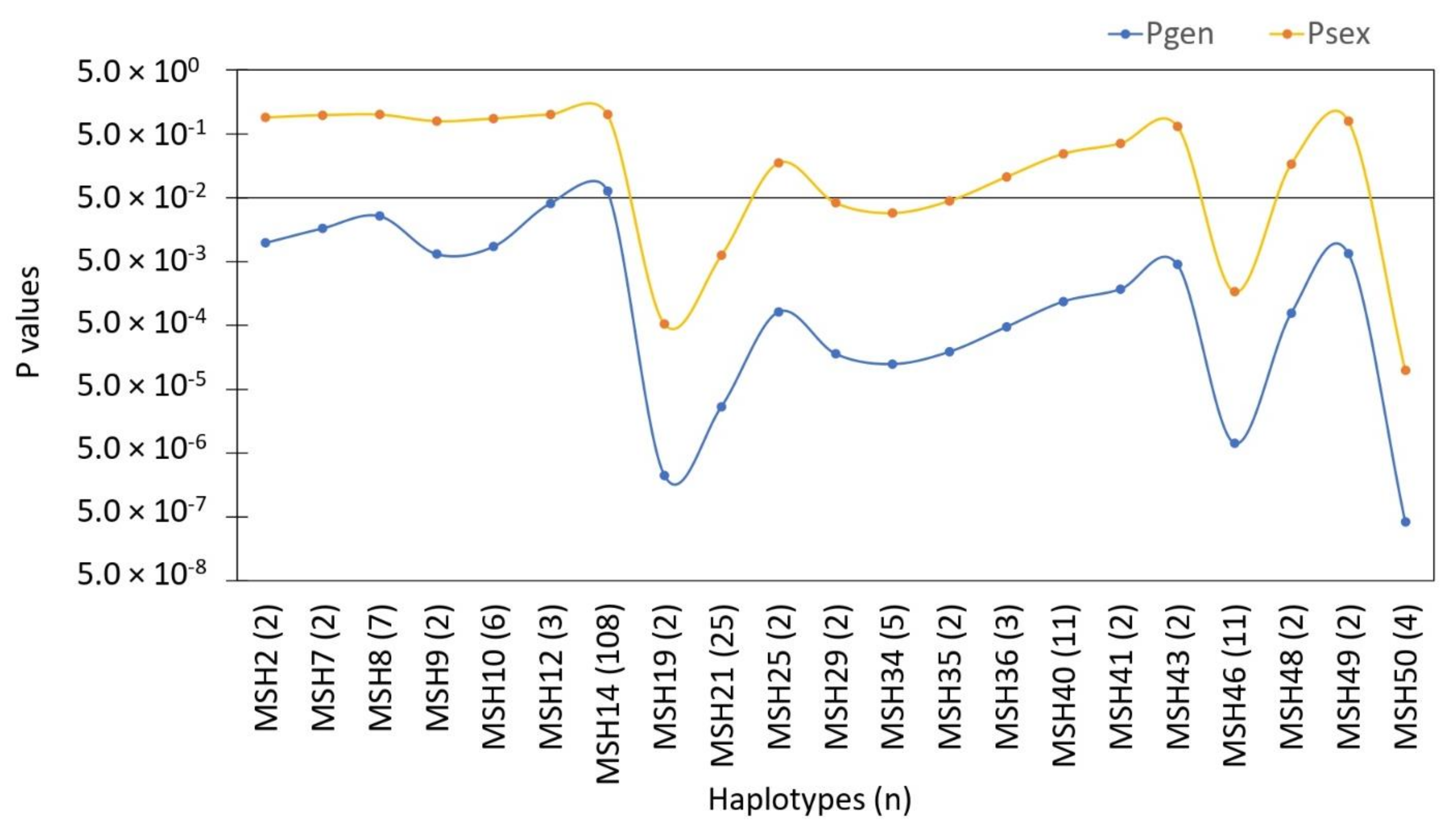
3. Results
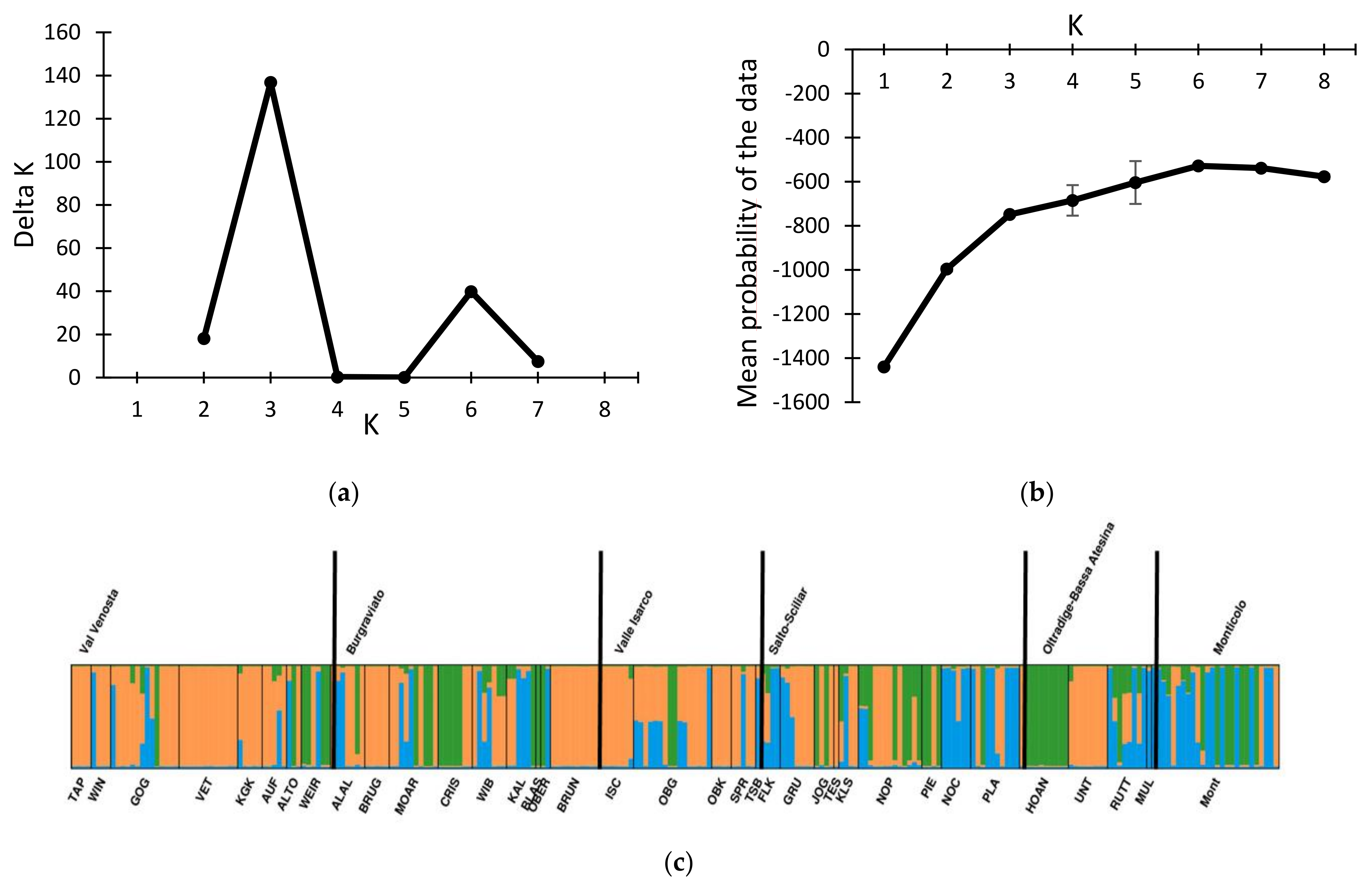
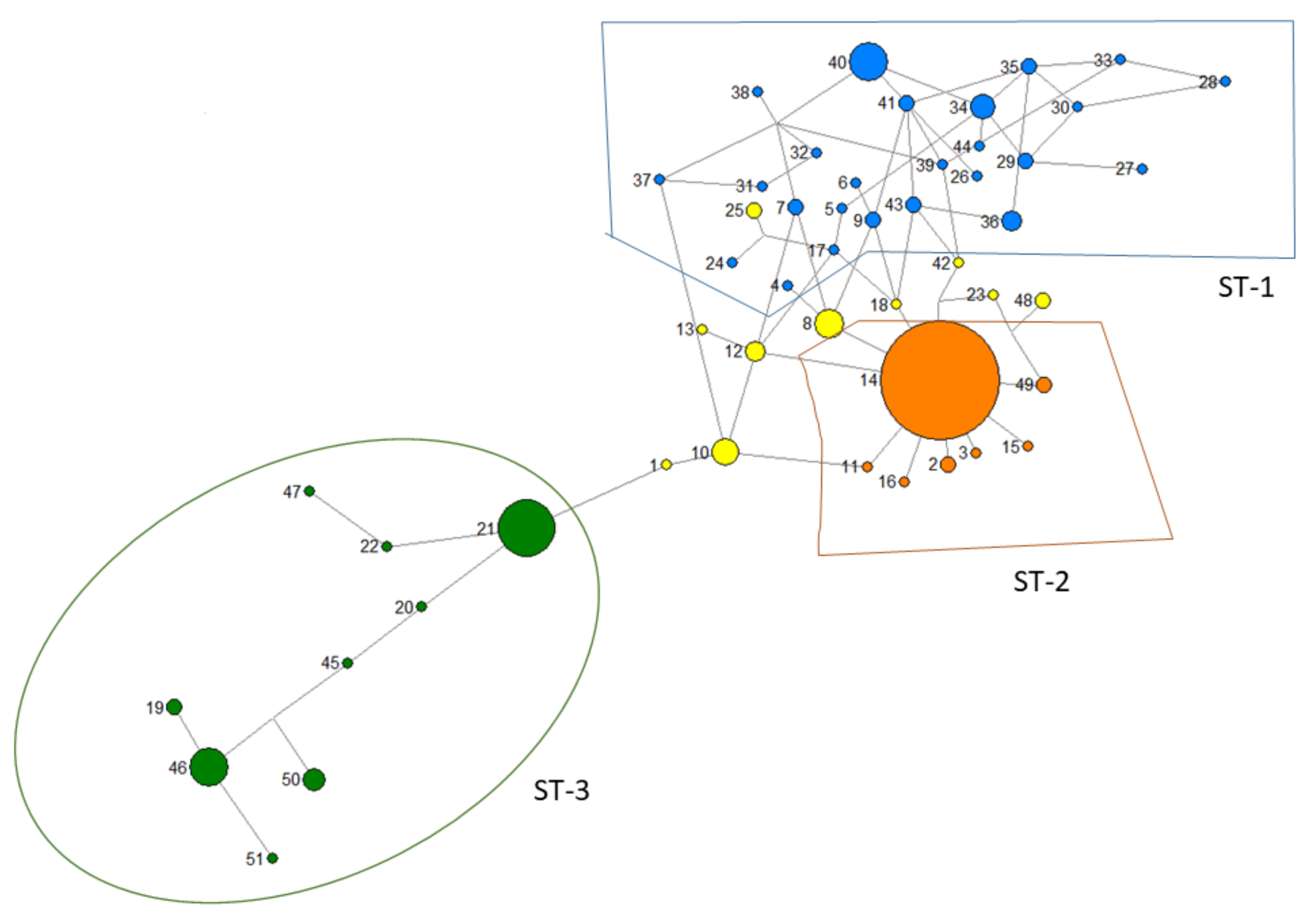
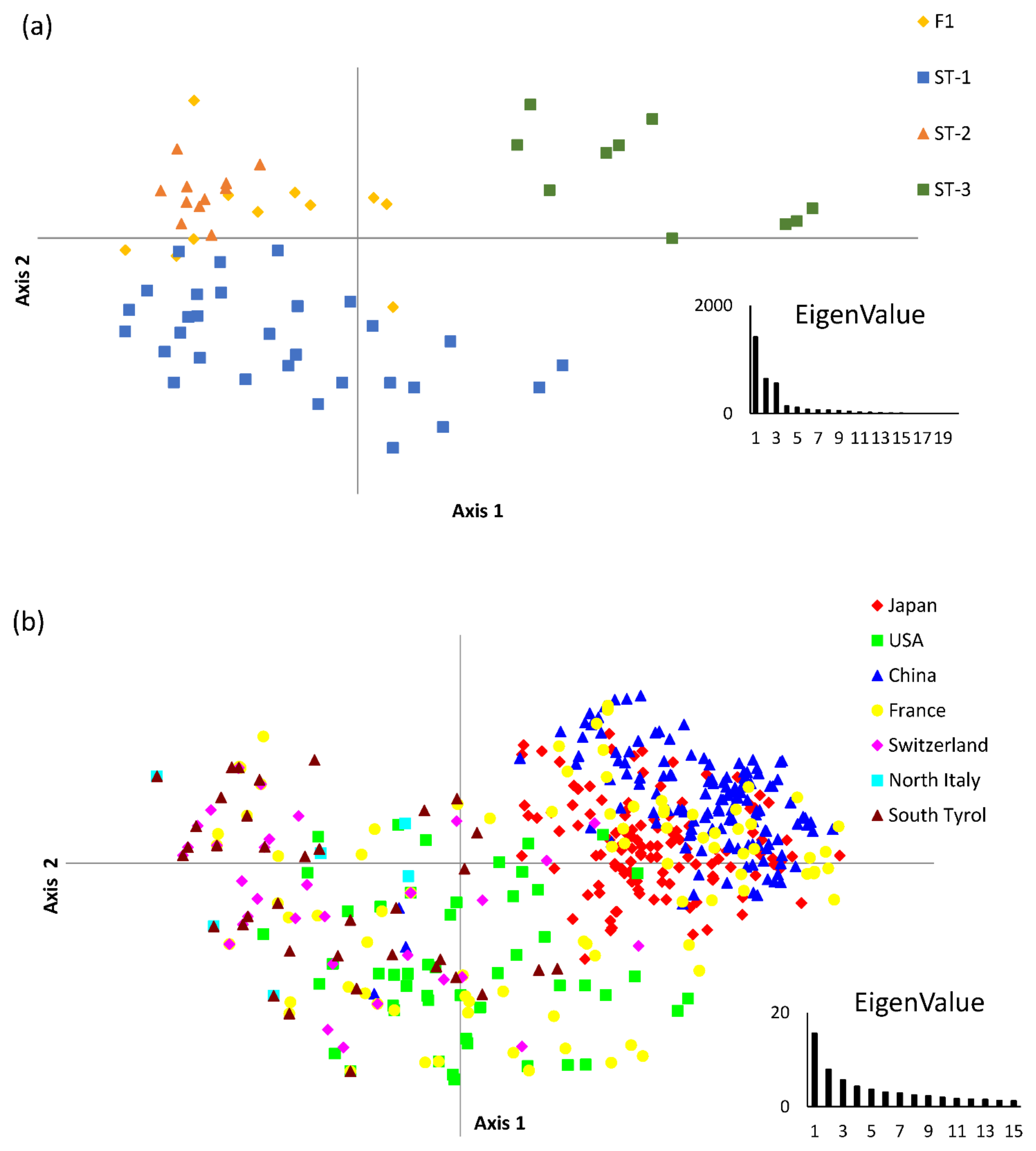
3.1. Genotypic Diversity and Population Structure
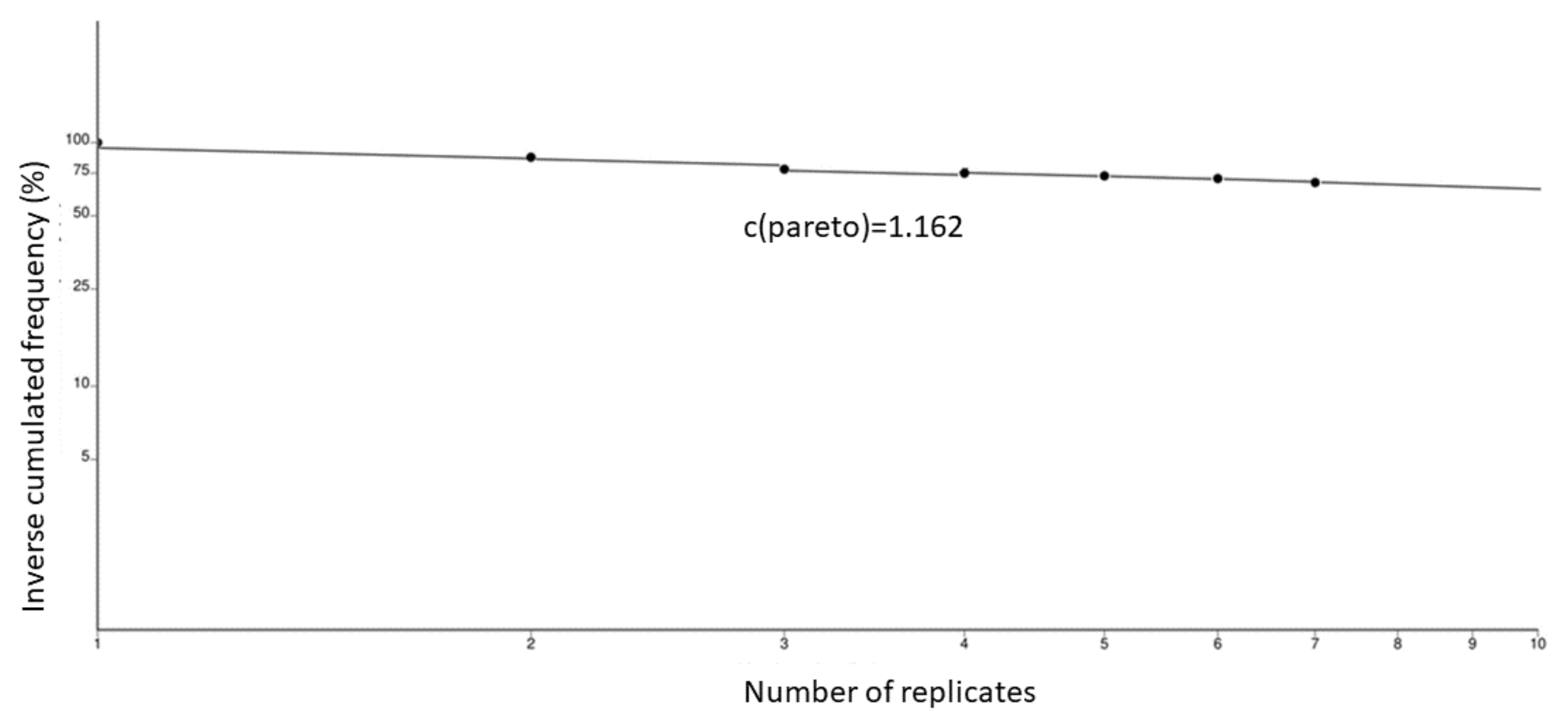
3.2. Selfing and Clonality
3.3. Relationship with Other Genetic Markers
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aukema, J.E.; Leung, B.; Kovacs, K.; Chivers, C.; Britton, K.O.; Englin, J.; Frankel, S.J.; Haight, R.G.; Holmes, T.P.; Liebhold, A.M. Economic impacts of non-native forest insects in the continental United States. PLoS ONE 2011, 6, e24587. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Ghelardini, L.; De Pace, C.; Desprez-Loustau, M.-L.; Capretti, P.; Chandelier, A.; Cech, T.; Chira, D.; Diamandis, S.; Gaitniekis, T.; et al. Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol. 2013, 197, 238–250. [Google Scholar] [CrossRef]
- Desprez-Loustau, M.; Robin, C.; Buee, M.; Courtecuisse, R.; Garbaye, J.; Suffert, F.; Sache, I.; Rizzo, D. The fungal dimension of biological invasions. Trends Ecol. Evol. 2007, 22, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, L.; Luchi, N.; Pecori, F.; Pepori, A.L.; Danti, R.; Della Rocca, G.; Capretti, P.; Tsopelas, P.; Santini, A. Ecology of invasive forest pathogens. Biol. Invasions 2017, 19, 3183–3200. [Google Scholar] [CrossRef]
- Anagnostakis, S. Chestnut blight: The classical problem of an introduced pathogen. Mycologia 1987, 79, 23. [Google Scholar] [CrossRef]
- Biraghi, A. La distribuzione del cancro del castagno in Italia. L’Italia For. E Mont. 1950, 5, 18–21. [Google Scholar]
- Heiniger, U.; Rigling, D. Biological control of chestnut blight in Europe. Annu. Rev. Phytopathol. 1994, 32, 581–599. [Google Scholar] [CrossRef]
- Peever, T.L.; Liu, Y.C.; Wang, K.; Hillman, B.I.; Foglia, R.; Milgroom, M.G. Incidence and diversity of double-stranded RNAs occurring in the chestnut blight fungus, Cryphonectria parasitica, in China and Japan. Phytopathology 1998, 88, 811–817. [Google Scholar] [CrossRef][Green Version]
- Berbegal, M.; Pérez-Sierra, A.; Armengol, J.; Grünwald, N.J. Evidence for multiple introductions and clonality in Spanish populations of Fusarium circinatum. Phytopathology 2013, 103, 851–861. [Google Scholar] [CrossRef]
- Goss, E.M.; Larsen, M.; Vercauteren, A.; Werres, S.; Heungens, K.; Grünwald, N.J. Phytophthora ramorum in Canada: Evidence for migration within North America and from Europe. Phytopathology 2011, 101, 166–171. [Google Scholar] [CrossRef]
- Dutech, C.; Fabreguettes, O.; Capdevielle, X.; Robin, C. Multiple introductions of divergent genetic lineages in an invasive fungal pathogen, Cryphonectria parasitica, in France. Heredity 2010, 105, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Dutech, C.; Barres, B.; Bridier, J.; Robin, C.; Milgroom, M.G.; Ravigné, V. The chestnut blight fungus world tour: Successive introduction events from diverse origins in an invasive plant fungal pathogen. Mol. Ecol. 2012, 21, 3931–3946. [Google Scholar] [CrossRef] [PubMed]
- Heiniger, U.; Rigling, D. Applicaction of the Cryphonectria hypovirus (CHV-1) to control the chestnut blight, experience from Switzerland. Acta Hortic. 2009, 815, 233–246. [Google Scholar] [CrossRef]
- Aguin, O.; Mata, M.; Mansilla, J.P.; Romero, A. Occurrence and diversity of vegetative compatibility types of Cryphonectria parasitica in Galicia (NW Spain). Acta Hortic. 2005, 693, 597–604. [Google Scholar] [CrossRef]
- Anagnostakis, S. Vegetative incompatibility in Endothia parasitica. Exp. Mycol. 1977, 1, 306–316. [Google Scholar] [CrossRef]
- Liu, Y.C.; Milgroom, M.G. High diversity of vegetative compatibility types in Cryphonectria parasitica in Japan and China. Mycologia 2007, 99, 279–284. [Google Scholar] [CrossRef]
- Mlinarec, J.; Ježić, M.; Ćosić, J.; Ćurković-Perica, M. Multilocus PCR assay reveals high diversity of vegetative compatibility types in populations of Cryphonectria parasitica in Croatia. Plant Pathol. 2018, 67, 741–749. [Google Scholar] [CrossRef]
- Moghadam, S.; Khodaparast, S.; Farsi, M. Vegetative compatibility groups in populations of Cryphonectria parasitica, the causal agent of chestnut blight in Guilan Province, Iran. Iran. J. Plant Pathol. 2010, 46, 1–3. [Google Scholar]
- Montenegro, D.; Aguín, O.; Sainz, M.J.; Hermida, M.; Mansilla, J.P. Diversity of vegetative compatibility types, distribution of mating types and occurrence of hypovirulence of Cryphonectria parasitica in chestnut stands in NW Spain. For. Ecol. Manag. 2008, 256, 973–980. [Google Scholar] [CrossRef]
- Murolo, S.; De Miccolis Angelini, R.M.; Faretra, F.; Romanazzi, G. Phenotypic and molecular investigations on hypovirulent Cryphonectria parasitica in Italy. Plant Dis. 2018, 102, 540–545. [Google Scholar] [CrossRef]
- Pérez-Sierra, A.; Romón-Ochoa, P.; Gorton, C.; Lewis, A.; Rees, H.; Van Der Linde, S.; Webber, J. High vegetative compatibility diversity of Cryphonectria parasitica infecting sweet chestnut (Castanea sativa) in Britain indicates multiple pathogen introductions. Plant Pathol. 2019, 68, 727–737. [Google Scholar] [CrossRef]
- Robin, C.; Capdevielle, X.; Martin, M.; Traver, C.; Colinas, C. Cryphonectria parasitica vegetative compatibility type analysis of populations in south-western France and northern Spain. Plant Pathol. 2009, 58, 527–535. [Google Scholar] [CrossRef]
- Short, D.P.G.; Double, M.; Nuss, D.L.; Stauder, C.M.; MacDonald, W.; Kasson, M.T. Multilocus PCR assays elucidate vegetative incompatibility gene profiles of Cryphonectria parasitica in the United States. Appl. Environ. Microbiol. 2015, 81, 5736–5742. [Google Scholar] [CrossRef] [PubMed]
- Sotirovski, K.; Papazova-Anakieva, I.; Grunwald, N.J.; Milgroom, M.G. Low diversity of vegetative compatibility types and mating type of Cryphonectria parasitica in the southern Balkans. Plant Pathol. 2004, 53, 325–333. [Google Scholar] [CrossRef]
- Trestic, T.; Uscuplic, M.; Colinas, C.; Rolland, G.; Giraud, A.; Robin, C. Vegetative compatibility type diversity of Cryphonectria parasitica populations in Bosnia-Herzegovina, Spain and France. For. Snow Landsc. Res. 2001, 76, 391–396. [Google Scholar]
- Bragança, H.; Simões, S.; Onofre, N.; Tenreiro, R.; Rigling, D. Cryphonectria parasitica in Portugal: Diversity of vegetative compatibility types, mating types, and occurrence of hypovirulence. For. Pathol. 2007, 37, 391–402. [Google Scholar] [CrossRef]
- Zamora, P.; Martín, A.B.; Rigling, D.; Diez, J.J. Diversity of Cryphonectria parasitica in western Spain and identification of hypovirus-infected isolates. For. Pathol. 2012, 42, 412–419. [Google Scholar] [CrossRef]
- Bryner, S.F.; Rigling, D. Hypovirus virulence and vegetative incompatibility in populations of the chestnut blight fungus. Phytopathology 2012, 102, 1161–1167. [Google Scholar] [CrossRef]
- Cornejo, C.; Šever, B.; Kupper, Q.; Prospero, S.; Rigling, D. A multiplexed genotyping assay to determine vegetative incompatibility and mating type in Cryphonectria parasitica. Eur. J. Plant Pathol. 2019, 155, 81–91. [Google Scholar] [CrossRef]
- Daldal, M.; Erincik, Ö.; Wall, J.R. Geographical distribution of vegetative compatibility and mating types of Cryphonectria parasitica in Izmir, Manisa and Denizli provinces in western Turkey. For. Pathol. 2018, 48, e12444. [Google Scholar] [CrossRef]
- Erincik, B.G.; Erincik, Ö.; Açikgöz, S. The potential diversity of vegetative compatibility types in the population of Cryphonectria parasitica in Turkey. For. Pathol. 2020, 50, e12646. [Google Scholar] [CrossRef]
- Ježić, M.; Kolp, M.; Prospero, S.; Sotirovski, K.; Double, M.; Rigling, D.; Risteski, M.; Karin-Kujundžić, V.; Idžojtić, M.; Poljak, I.; et al. Diversity of Cryphonectria parasitica in callused chestnut blight cankers on European and American chestnut. For. Pathol. 2019, 49, e12566. [Google Scholar] [CrossRef]
- Ježić, M.; Schwarz, J.M.; Prospero, S.; Sotirovski, K.; Risteski, M.; Ćurković-Perica, M.; Nuskern, L.; Krstin, L.; Katanić, Z.; Maleničić, E.; et al. Temporal and spatial genetic population structure of Cryphonectria parasitica and its associated hypovirus across an invasive range of chestnut blight in Europe. Phytopathology 2021, 111, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Krstin, L.; Novak-Agbaba, S.; Rigling, D.; Krajačić, M.; Ćurković Perica, M. Chestnut blight fungus in Croatia: Diversity of vegetative compatibility types, mating types and genetic variability of associated Cryphonectria hypovirus 1. Plant Pathol. 2008, 57, 1086–1096. [Google Scholar] [CrossRef]
- Cortesi, P.; Rigling, D.; Heiniger, U. Comparison of vegetative compatibility types in Italian and Swiss subpopulations of Cryphonectria parasitica. Eur. J. For. Pathol. 1998, 28, 167–176. [Google Scholar] [CrossRef]
- Pearson, M.N.; Beever, R.E.; Boine, B.; Arthur, K. Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol. Plant Pathol. 2009, 10, 115–128. [Google Scholar] [CrossRef]
- Breuillin, F.; Dutech, C.; Robin, C. Genetic diversity of the chestnut blight fungus Cryphonectria parasitica in four French populations assessed by microsatellite markers. Mycol. Res. 2006, 110, 288–296. [Google Scholar] [CrossRef]
- Jarne, P.; Lagoda, P.J.L. Microsatellites, from molecules to populations and back. Trends Ecol. Evol. 1996, 11, 424–429. [Google Scholar] [CrossRef]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef]
- Giraud, T.; Enjalbert, J.; Fournier, E.; Delmotte, F.; Dutech, C. Population genetics of fungal diseases of plants. Parasite 2008, 15, 449–454. [Google Scholar] [CrossRef]
- Ahmad, F.; Baric, S. Genetic diversity of Cryphonectria parasitica causing chestnut blight in South Tyrol (northern Italy). Eur. J. Plant Pathol. 2022, 162, 621–635. [Google Scholar] [CrossRef]
- Cassago, A.; Panepucci, R.A.; Baião, A.M.T.; Henrique-Silva, F. Cellophane based mini-prep method for DNA extraction from the filamentous fungus Trichoderma Reesei. BMC Microbiol. 2002, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Kubisiak, T.L.; Dutech, C.; Milgroom, M.G. Fifty-three polymorphic microsatellite loci in the chestnut blight fungus, Cryphonectria parasitica. Mol. Ecol. Notes 2007, 7, 428–432. [Google Scholar] [CrossRef]
- Davis, J.E.; Kubisiak, T.L.; Milgroom, M.G. Polymorphic sequence-characterized codominant loci in the chestnut blight fungus, Cryphonectria parasitica. Mol. Ecol. Notes 2005, 5, 195–197. [Google Scholar] [CrossRef]
- Peakall, R.O.D.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Eliades, N.G.; Eliades, D.G. HAPLOTYPE ANALYSIS: Software for analysis of haplotype data. Distributed by the authors. In Forest Genetics and Forest Tree Breeding; Georg-Augst University Göttingen: Göttingen, Germany, 2009. [Google Scholar]
- Arnaud-Haond, S.; Belkhir, K. GENCLONE: A computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol. Ecol. Notes 2007, 7, 15–17. [Google Scholar] [CrossRef]
- Wang, X.R.; Szmidt, A.E.; Ennos, R.A.; Hansson, P. Genetic variability in the canker pathogen fungus, Gremmeniella abietina. 2. Fine-scale investigation of the population genetic structure. Can. J. Bot. 1997, 75, 1460–1469. [Google Scholar] [CrossRef]
- Agapow, P.M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Dutech, C.; Rossi, J.-P.; Fabreguettes, O.; Robin, C. Geostatistical genetic analysis for inferring the dispersal pattern of a partially clonal species: Example of the chestnut blight fungus. Mol. Ecol. 2008, 17, 4597–4607. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Prospero, S.; Rigling, D. Invasion genetics of the chestnut blight fungus Cryphonectria parasitica in Switzerland. Phytopathology 2012, 102, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Rostagno, L. Interaction between Cryphonectria parasitica and CHV1: A Model for Virus Caused Hypovirulence. Ph.D. Thesis, Università degli Studi di Torino, Turin, Italy, 2009. [Google Scholar]
- Baric, S.; Storti, A.; Hofer, M.; Guerra, W.; Dalla Via, J. Molecular genetic identification of apple cultivars based on microsatellite DNA analysis. I. The database of 600 validated profiles. Erwerbs-Obstbau 2020, 62, 117–154. [Google Scholar] [CrossRef]
- Kivelä, M.; Arnaud-Haond, S.; Saramäki, J. EDENetworks: A user-friendly software to build and analyse networks in biogeography, ecology and population genetics. Mol. Ecol. Resour. 2015, 15, 117–122. [Google Scholar] [CrossRef]
- Grünwald, N.J.; Goss, E.M. Evolution and population genetics of exotic and re-emerging pathogens: Novel tools and approaches. Annu. Rev. Phytopathol. 2011, 49, 249–267. [Google Scholar] [CrossRef]
- Milgroom, M.G.; Sotirovski, K.; Spica, D.; Davis, J.E.; Brewer, M.T.; Milev, M.; Cortesi, P. Clonal population structure of the chestnut blight fungus in expanding ranges in southeastern Europe. Mol. Ecol. 2008, 17, 4446–4458. [Google Scholar] [CrossRef]
- Stauber, L.; Badet, T.; Feurtey, A.; Prospero, S.; Croll, D. Emergence and diversification of a highly invasive chestnut pathogen lineage across southeastern Europe. eLife 2021, 10, e56279. [Google Scholar] [CrossRef]
- Maresi, G.; Minerbi, S.; Sottovia, A.; Turchetti, T. Der Kastanienrinderkrebs in Südtirol. Allg. Forst Z. Waldwirtsch. Umweltvorsorge 1993, 3, 140–144. [Google Scholar]
- Windegger, A. La Lotta Biologica Contro il Cancro del Castagno in Alto Adige. Bachelor’s Thesis, The University of Firenze, Florence, Italy, 1994. [Google Scholar]
- Nuss, D.L. Hypovirulence: Mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 2005, 3, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Zamora, P.; Rigling, D.; Diez, J.J. Detection of hypovirulent isolates of Cryphonectria parasitica in Castilla y Leon, Spain. Acta Hortic. 2008, 784, 163–168. [Google Scholar] [CrossRef]
- Castaño, C.; Bassie, L.; Oliach, D.; Gómez, M.; Medina, V.; Liu, B.; Colinas, C. Cryphonectria hypovirus 1 (CHV-1) survey reveals low occurrence and diversity of subtypes in NE Spain. For. Pathol. 2014, 45, 51–59. [Google Scholar] [CrossRef]
- Peters, F.S.; Bußkamp, J.; Prospero, S.; Rigling, D.; Metzler, B. Genetic diversification of the chestnut blight fungus Cryphonectria parasitica and its associated hypovirus in Germany. Fungal Biol. 2014, 118, 193–210. [Google Scholar] [CrossRef]
- Peever, T.L.; Liu, Y.C.; Cortesi, P.; Milgroom, M.G. Variation in tolerance and virulence in the chestnut blight fungus-hypovirus interaction. Appl. Environ. Microbiol. 2000, 66, 4863–4869. [Google Scholar] [CrossRef]
- Demené, A.; Legrand, L.; Gouzy, J.; Debuchy, R.; Saint-Jean, G.; Fabreguettes, O.; Dutech, C. Whole-genome sequencing reveals recent and frequent genetic recombination between clonal lineages of Cryphonectria parasitica in western Europe. Fungal Genet. Biol. 2019, 130, 122–133. [Google Scholar] [CrossRef]
- Milgroom, M.G.; del Mar Jiménez-Gasco, M.; Garcia, C.O.; Drott, M.T.; Jiménez-Diaz, R.M. Recombination between clonal lineages of the asexual fungus Verticillium dahliae detected by genotyping by sequencing. PLoS ONE 2014, 9, e106740. [Google Scholar] [CrossRef]
- Henk, D.A.; Shahar-Golan, R.; Devi, K.R.; Boyce, K.J.; Zhan, N.; Fedorova, N.D.; Nierman, W.C.; Hsueh, P.-R.; Yuen, K.-Y.; Sieu, T.P.M.; et al. Clonality despite sex: The evolution of host-associated sexual neighborhoods in the pathogenic fungus Penicillium marneffei. PLoS Pathog. 2012, 8, e1002851. [Google Scholar] [CrossRef]
- Romon-Ochoa, P.; Kranjec Orlović, J.; Gorton, C.; Lewis, A.; van der Linde, S.; Pérez-Sierra, A. New detections of chestnut blight in Britain during 2019–2020 reveal high Cryphonectria parasitica diversity and limited spread of the disease. Plant Pathol. 2021. [Google Scholar] [CrossRef]
- Associazione Turistica Lana e Dintorni. L’uomo Che Sussurrava… Alle Castagne. Available online: https://www.merano-suedtirol.it/it/lana-e-dintorni/informazioni-servizi/brochures/greatthings/l-uomo-che-sussurrava-alle-castagne.html (accessed on 1 October 2021).
- Mair, K. Die Esskastanien-Gallwespe–ein neuer, gefährlicher Schädling. Obstbau Weinbau 2009, 46, 149–150. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′ to 3′) a | Conc. [µM] b | Size Range (bp) c | NA d | Nucleotide Motif | Multiplex Reaction | Reference |
|---|---|---|---|---|---|---|---|
| CPE1-F | 6-FAM-GTCTCACCACACATCGCAAG | 0.4 0.4 | 127–145 | 2 | (GT)n | 3 | [37] |
| CPE1-R | TCATCACGACAAAAGAAGACG | ||||||
| CPE3-F | 6-FAM-CAACTCGTCACCCACCTTG | 0.2 | 188–190 | 2 | (GT)n | 1 | [37] |
| CPE3-R | ATAGGCATTCTCCGCTCTTG | 0.2 | |||||
| CPE5-F | HEX-TGTCAACAACGGTCAACACC | 0.2 0.2 | 248–257 | 3 | (AAC)n | 3 | [37] |
| CPE5-R | CGGAGAGGAGAACTCTGAGAC | ||||||
| CPE8-F | 6-FAM-GCGAGCAAGCGTGATTCC | 0.4 0.4 | 109–115 | 2 | (AAC)n | 2 | [37] |
| CPE8-R | GCTCGTCCTGGTCCTCCT | ||||||
| CPG3-F | HEX-CGTAAGGCAGAGGCAGAGAC | 0.2 0.2 | 194–212 | 3 | (GT)n | 2 | [37] |
| CPG3-R | TCCCTATGCCCAAGACACTC | ||||||
| CPG4-F | 6-FAM-ATGCTCCGAAGGTTTGTCAC | 0.2 0.2 | 186–206 | 3 | (GA)n | 3 | [37] |
| CPG4-R | CAACCGCAATCTGTTTCCTC | ||||||
| CPG6-F | 6-FAM-ATCATCACGACGCAATGGTA | 0.2 0.2 | 240–262 | 3 | (GA)n | 1 | [43] |
| CPG6-R | TCCGGGCATTCAGCAMAT | ||||||
| CPG14-F | 6-FAM-TTCTGAAGGTGGTTGTGGTG | 0.2 0.2 | 252–264 | 2 | (AGGAAG)n | 3 | [43] |
| CPG14-R | GGTCCGAACCATCAAAAGAC | ||||||
| CPSI006-F | HEX-ATGTCGAGTTTACCCGATGG | 0.2 0.2 | 133–139 | 3 | (AC)n | 1 e | [43] |
| CPSI006-R | GAGATGTGTGGAATGCAACG | ||||||
| CPSI009-F | 6-FAM-ATCATCCATCCTGTCCGAGT | 0.2 0.2 | 138–156 | 4 | (AT)n | 2 e | [43] |
| CPSI009-R | TGGGGTTGGCATAATCTTCT | ||||||
| IO7-650-F | 6-FAM-CATGCGAGAAATGCAGGAGTGTTG | 0.2 0.2 | 270–290 | 3 | (AC)n | 2 | [44] |
| IO7-650-R | GGGCTCCAGGATATCGAAGACATT |
| SSR Locus | Switzerland [56] | France [11] | Northern Italy [57] | China, Japan and USA [12] |
|---|---|---|---|---|
| CPE1 | −3 | −2 | −2 | −2 |
| CPE3 | −4 | −4 | −4 | −4 |
| CPE5 | −4 | −3 | −3 | −3 |
| CPE8 | −2 | −3 | −3 | −3 |
| CPG3 | −4 | −2 | −2 | −2 |
| CPG4 | −3 | −1 | −1 | −1 |
| CPG6 | −3 | −2 | −2 | −2 |
| CPG14 | −4 | −3 | −3 | −3 |
| IO7-650 | −4 | - | - | - |
| CPSI006 | - | - | −3 | - |
| CPSI009 | - | - | −4 | - |
| District a | Chestnut Stand b | N. Isolates | N. Haplotypes | Dominant Haplotype c |
|---|---|---|---|---|
| Val Venosta–Vinschgau | TAP | 4 | 1 | - |
| WIN | 4 | 2 | - | |
| GOG | 14 | 7 | MSH14 | |
| VET | 12 | 4 | MSH14 | |
| KGK | 5 | 2 | MSH14 | |
| AUF | 5 | 3 | MSH14 | |
| ALTO | 3 | 3 | - | |
| WEIR | 5 | 3 | * | |
| Burgraviato–Burggrafenamt | ALAL | 7 | 4 | MSH14 |
| BRUG | 5 | 1 | MSH14 | |
| MOAR | 10 | 6 | * | |
| CRIS | 7 | 2 | MSH21 | |
| WIB | 7 | 5 | * | |
| KAL | 6 | 5 | * | |
| BLAS | 1 | 1 | - | |
| OBER | 2 | 2 | - | |
| BRUN | 10 | 1 | MSH14 | |
| Valle Isarco–Eisacktal | ISC | 7 | 4 | MSH14 |
| OBG | 14 | 4 | * | |
| OBK | 4 | 1 | MSH14 | |
| SPR | 5 | 2 | MSH14 | |
| TSB | 1 | 1 | - | |
| Salto-Sciliar–Salten-Schlern | FLK | 4 | 3 | - |
| GRU | 7 | 4 | MSH14 | |
| JOG | 3 | 2 | - | |
| TES | 1 | 1 | - | |
| KLS | 4 | 3 | - | |
| NOP | 13 | 3 | * | |
| PIE | 3 | 2 | - | |
| NOC | 4 | 2 | - | |
| PLA | 9 | 4 | MSH40 | |
| Oltradige-Bassa Atesina–Überetsch-Unterland | HOAN | 10 | 3 | MSH21 |
| UNT | 8 | 1 | MSH14 | |
| RUTT | 7 | 4 | * | |
| MUL | 1 | 1 | - | |
| Monticolo Forest | Mont | 23 | 12 | MSH11 |
| Microsatellite Haplotype | n | C a | MAT-1 b | MAT-2 b | Dominant VC Type c | % of Dominant VC Type d | Haplotype ITS | ||
|---|---|---|---|---|---|---|---|---|---|
| H1 | H2 | H3 | |||||||
| MSH14 | 108 | ST-2 | 68 | 35 | EU-2 | 84.1 | 1 | 16 | 1 |
| MSH21 | 25 | ST-3 | 3 | 22 | EU-13 | 80.0 | 3 | 0 | 0 |
| MSH40 | 11 | ST-1 | 3 | 7 | EU-5 | 54.6 | 1 | 0 | 1 |
| MSH46 | 11 | ST-3 | 7 | 4 | EU-17 | 90.9 | 1 | 0 | 0 |
| MSH8 | 7 | ST-1×2 * | 1 | 5 | EU-2 | 83.3 | 0 | 2 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, F.; Baric, S. Microsatellite Analysis Revealing High Genetic Diversity of the Chestnut Blight Fungus in South Tyrol (Northern Italy). Forests 2022, 13, 344. https://doi.org/10.3390/f13020344
Ahmad F, Baric S. Microsatellite Analysis Revealing High Genetic Diversity of the Chestnut Blight Fungus in South Tyrol (Northern Italy). Forests. 2022; 13(2):344. https://doi.org/10.3390/f13020344
Chicago/Turabian StyleAhmad, Farooq, and Sanja Baric. 2022. "Microsatellite Analysis Revealing High Genetic Diversity of the Chestnut Blight Fungus in South Tyrol (Northern Italy)" Forests 13, no. 2: 344. https://doi.org/10.3390/f13020344
APA StyleAhmad, F., & Baric, S. (2022). Microsatellite Analysis Revealing High Genetic Diversity of the Chestnut Blight Fungus in South Tyrol (Northern Italy). Forests, 13(2), 344. https://doi.org/10.3390/f13020344