
Genome-Wide Variation in DNA Methylation Predicts Variation in Leaf Traits in an Ecosystem-Foundational Oak Species

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Species
2.2. Valley Oak Provenance Trial
2.3. Phenotypic Measurements
2.4. Bisulfite Sequencing
2.5. Methylation and SNP Calling
2.6. Trait Associations
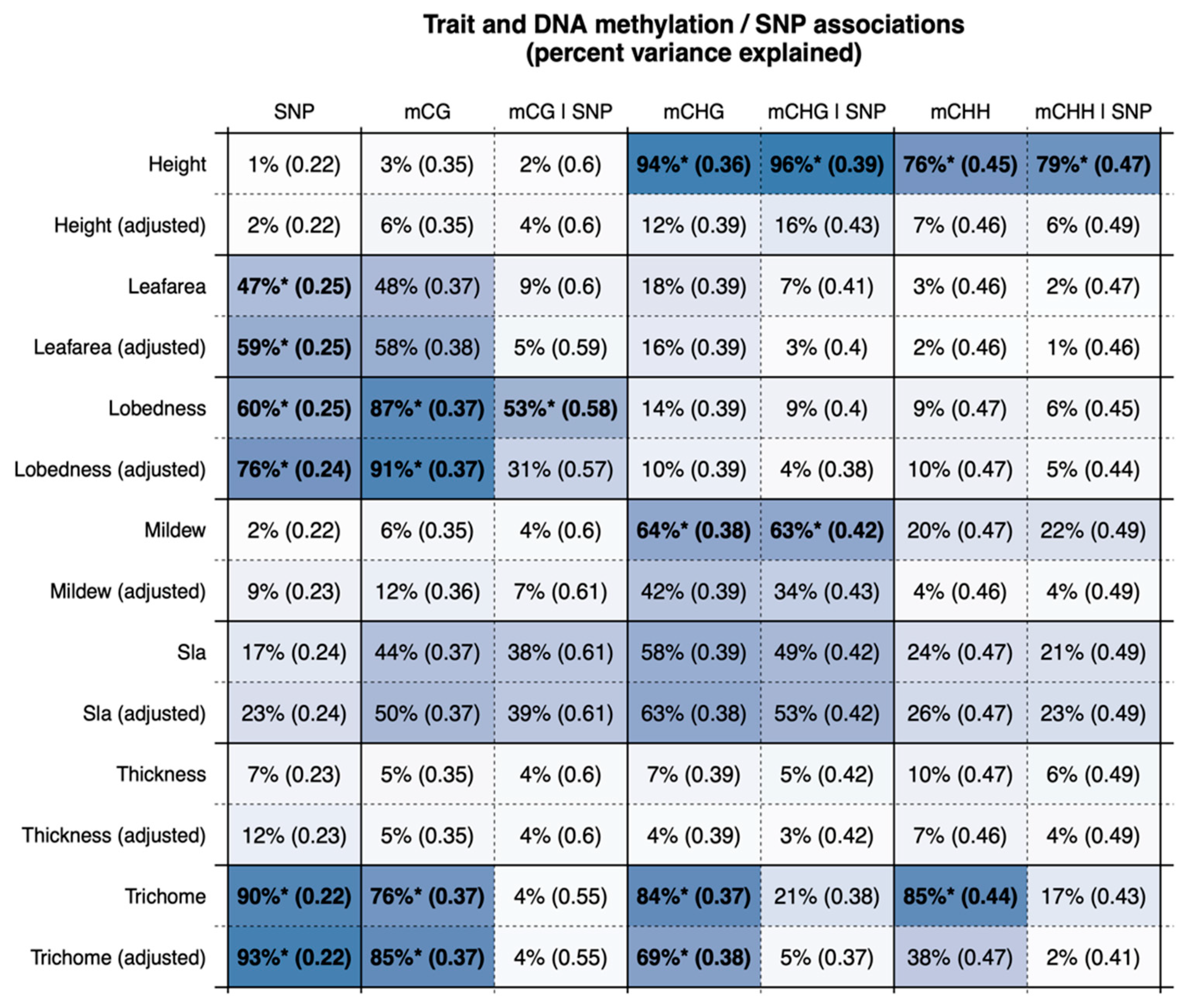
3. Results
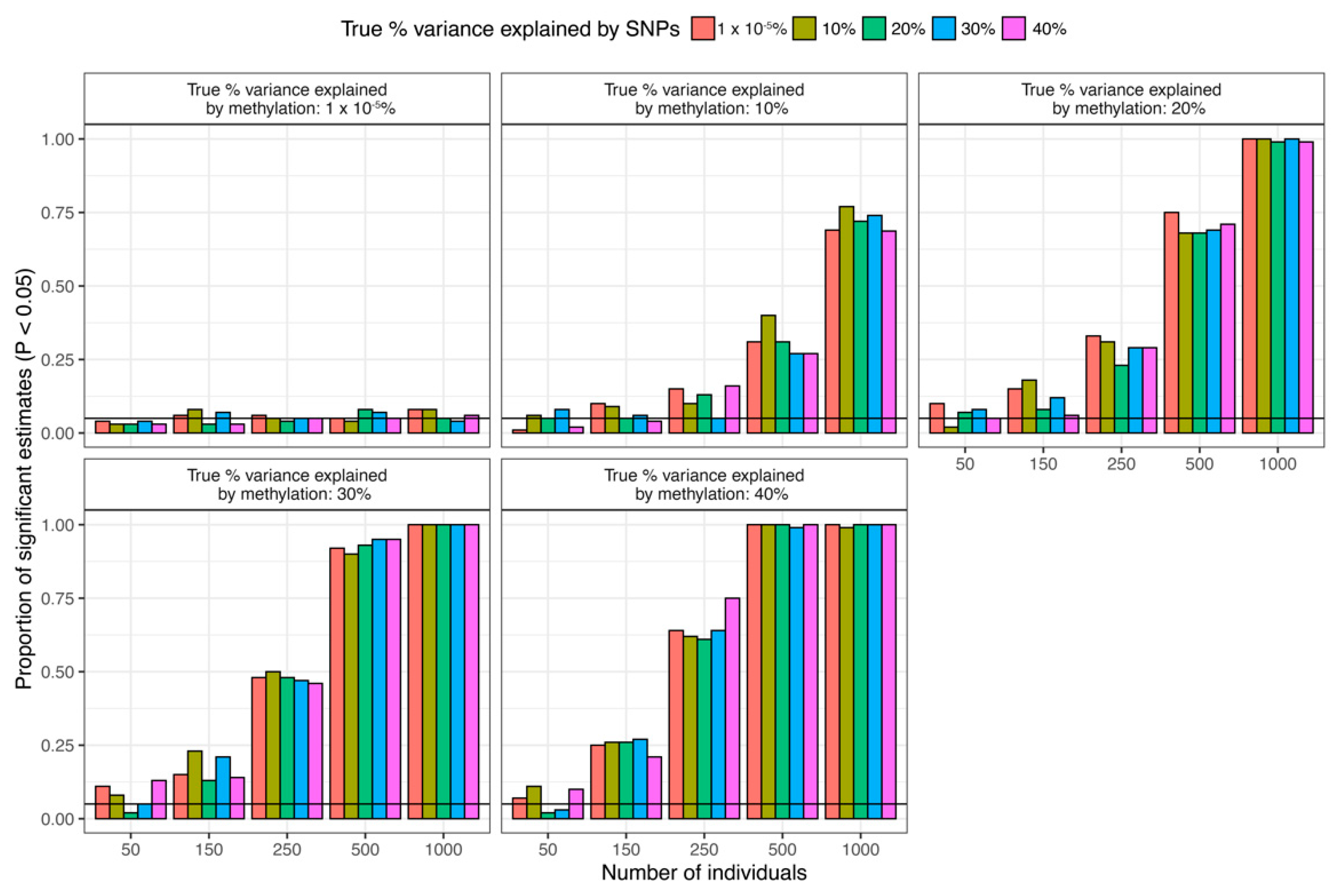
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sow, M.D.; Allona, I.; Ambroise, C.; Conde, D.; Fichot, R.; Gribkova, S.; Jorge, V.; Le-Provost, G.; Pâques, L.; Plomion, C.; et al. Epigenetics in forest trees: State of the art and potential implications for breeding and management in a context of climate change. In Advances in Botanical Research; Mirouze, M., Bucher, E., Gallusci, P., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 88, pp. 387–453. [Google Scholar]
- Carbó, M.; Iturra, C.; Correia, B.; Colina, F.J.; Meijón, M.; Álvarez, J.M.; Cañal, M.J.; Hasbún, R.; Pinto, G.; Valledor, L. Epigenetics in forest trees: Keep calm and carry on. In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications: Transcriptional Regulation and Chromatin Remodelling in Plants; Alvarez-Venegas, R., De-la-Peña, C., Casas-Mollano, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 381–403. ISBN 9783030147600. [Google Scholar]
- Balao, F.; Paun, O.; Alonso, C. Uncovering the contribution of epigenetics to plant phenotypic variation in Mediterranean ecosystems. Plant Biol. 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.L.; Alonso, C.; Becker, C.; Bossdorf, O.; Bucher, E.; Colomé-Tatché, M.; Durka, W.; Engelhardt, J.; Gaspar, B.; Gogol-Döring, A.; et al. Ecological plant epigenetics: Evidence from model and non-model species, and the way forward. Ecol. Lett. 2017, 20, 1576–1590. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; VonHoldt, B.M.; Sork, V.L. Epigenetics in ecology and evolution: What we know and what we need to know. Mol. Ecol. 2016, 25, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Genger, R.K.; Peacock, W.J.; Dennis, E.S. DNA methylation in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establising, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2011, 11, 204–220. [Google Scholar] [CrossRef]
- Bossdorf, O.; Richards, C.L.; Pigliucci, M. Epigenetics for ecologists. Ecol. Lett. 2008, 11, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.F.; Jansen, J.J.; van Dijk, P.J.; Biere, A. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010, 185, 1108–1118. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Fischer, M.; Colot, V.; Bossdorf, O. Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 2013, 197, 314–322. [Google Scholar] [CrossRef]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef]
- Herrera, C.M.; Alonso, C.; Medrano, M.; Pérez, R.; Bazaga, P. Transgenerational epigenetics: Inheritance of global cytosine methylation and methylation-related epigenetic markers in the shrub Lavandula latifolia. Am. J. Bot. 2018, 105, 741–748. [Google Scholar] [CrossRef]
- Herman, J.J.; Sultan, S.E. DNA methylation mediates genetic variation for adaptive transgenerational plasticity. Proc. Royal Soc. B 2016, 283, 20160988. [Google Scholar] [CrossRef] [PubMed]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-colombani, V.; Albuisson, J.; Heredia, F.; Colot, V. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Whipple, A.V.; Holeski, L.M. Epigenetic inheritance across the landscape. Front. Genet. 2016, 7, 189. [Google Scholar] [CrossRef]
- Zhang, Y.; Wendte, J.M.; Ji, L.; Schmitz, R.J. Natural variation in DNA methylation homeostasis and the emergence of epialleles. Proc. Natl. Acad. Sci. USA 2020, 117, 4874–4884. [Google Scholar] [CrossRef]
- Fieldes, M.A.; Amyot, L.M. Epigenetic control of early flowering in flax lines induced by 5- azacytidine applied to germinating seed. J. Hered. 1999, 90, 199–206. [Google Scholar] [CrossRef][Green Version]
- Dubin, M.J.; Zhang, P.; Meng, D.; Remigereau, M.S.; Osborne, E.J.; Casale, F.P.; Drewe, P.; Kahles, A.; Jean, G.; Vilhjálmsson, B.; et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 2015, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef]
- Tatra, G.S.; Miranda, J.; Chinnappa, C.C.; Reid, D.M. Effect of light quality and 5-azacytidine on genomic methylation and stem elongation in two ecotypes of Stellaria longipes. Physiol. Plant. 2000, 109, 313–321. [Google Scholar] [CrossRef]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluch, S.; Fraga, M.F.; Guevara, M.Á.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef]
- Brockerhoff, E.G.; Barbaro, L.; Castagneyrol, B.; Forrester, D.I.; Gardiner, B.; González-Olabarria, J.R.; Lyver, P.O.; Meurisse, N.; Oxbrough, A.; Taki, H.; et al. Forest biodiversity, ecosystem functioning and the provision of ecosystem services. Biodivers. Conserv. 2017, 26, 3005–3035. [Google Scholar] [CrossRef]
- Bonan, G.B. Forests and climate change: Forcings, feedbacks, and the climate benefits of forests. Science 2008, 320, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Beer, C.; Reichstein, M.; Tomelleri, E.; Ciais, P.; Jung, M.; Carvalhais, N.; Rödenbeck, C.; Arain, M.A.; Baldocchi, D.; Bonan, G.B.; et al. Terrestrial gross carbon dioxide uptake: Global distribution and covariation with climate. Science 2010, 329, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Cavender-Bares, J. Diversity, distribution and ecosystem services of the North American oaks. Int. Oaks 2016, 27, 37–48. [Google Scholar]
- Platt, A.; Gugger, P.F.; Pellegrini, M.; Sork, V.L. Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol. Ecol. 2015, 24, 3823–3830. [Google Scholar] [CrossRef]
- Gugger, P.F.; Fitz-Gibbon, S.; Pellegrini, M.; Sork, V.L. Species-wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol. Ecol. 2016, 25, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Rico, L.; Ogaya, R.; Barbeta, A.; Peñuelas, J. Changes in DNA methylation fingerprint of Quercus ilex trees in response to experimental field drought simulating projected climate change. Plant Biol. 2014, 16, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Correia, B.; Valledor, L.; Meijón, M.; Rodriguez, J.L.; Dias, M.C.; Santos, C.; Cañal, M.J.; Rodriguez, R.; Pinto, G. Is the interplay between epigenetic markers related to the acclimation of cork oak plants to high temperatures? PLoS ONE 2013, 8, e53543. [Google Scholar] [CrossRef]
- Ramos, M.; Rocheta, M.; Carvalho, L.; Inácio, V.; Graça, J.; Morais-Cecilio, L. Expression of DNA methyltransferases is involved in Quercus suber cork quality. Tree Genet. Genomes 2013, 9, 1481–1492. [Google Scholar] [CrossRef][Green Version]
- Browne, L.; Mead, A.; Horn, C.; Chang, K.; Celikkol, Z.A.; Henriquez, C.L.; Ma, F.; Beraut, E.; Meyer, R.S.; Sork, V.L. Experimental DNA demethylation associates with changes in growth and gene expression of oak tree seedlings. G3 2020, 10, 1019–1028. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Asante, D.K.A.; Fossdal, C.G.; Junttila, O.; Johnsen, Ø. Differential gene expression related to an epigenetic memory affecting climatic adaptation in Norway spruce. Plant Sci. 2011, 180, 132–139. [Google Scholar] [CrossRef]
- Liang, D.; Zhang, Z.; Wu, H.; Huang, C.; Shuai, P.; Ye, C.-Y.; Tang, S.; Wang, Y.; Yang, L.; Wang, J.; et al. Single-base-resolution methylomes of Populus trichocarpa reveal the association between DNA methylation and drought stress. BMC Genet. 2014, 15, S9. [Google Scholar] [CrossRef]
- Jacinto Pereira, W.; de Castro Rodrigues Pappas, M.; Camargo Campoe, O.; Stape, J.L.; Grattapaglia, D.; Joannis Pappas, G., Jr. Patterns of DNA methylation changes in elite Eucalyptus clones across contrasting environments. For. Ecol. Manag. 2020, 474, 118319. [Google Scholar] [CrossRef]
- Albaladejo, R.G.; Parejo-Farnés, C.; Rubio-Pérez, E.; Nora, S.; Aparicio, A. Linking DNA methylation with performance in a woody plant species. Tree Genet. Genomes 2019, 15, 15. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linn. Soc. 2013, 171, 441–452. [Google Scholar] [CrossRef]
- Kalisz, S.; Purugganan, M.D. Epialleles via DNA methylation: Consequences for plant evolution. Trends Ecol. Evol. 2004, 19, 309–314. [Google Scholar] [CrossRef]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA methylation in context: From genomes to gene expression in plants. Biochim. Biophys. Acta 2017, 1860, 149–156. [Google Scholar] [CrossRef]
- Bewick, A.J.; Zhang, Y.; Wendte, J.M.; Zhang, X.; Schmitz, R.J. Evolutionary and experimental loss of gene body methylation and its consequence to gene expression. G3 2019, 9, 2441–2445. [Google Scholar] [CrossRef]
- Kundariya, H.; Yang, X.; Morton, K.; Sanchez, R.; Axtell, M.J.; Hutton, S.F.; Fromm, M.; Mackenzie, S.A. MSH1-induced heritable enhanced growth vigor through grafting is associated with the RdDM pathway in plants. Nat. Commun. 2020, 11, 5343. [Google Scholar] [CrossRef]
- Gienapp, P.; Fior, S.; Guillaume, F.; Lasky, J.R.; Sork, V.L.; Csilléry, K. Genomic quantitative genetics to study evolution in the wild. Trends Ecol. Evol. 2017, 32, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, M.; Dugas, E.; Benchouaia, M.; Leduque, B.; Jiménez-Gómez, J.M.; Colot, V.; Quadrana, L. The impact of transposable elements on tomato diversity. Nat. Commun. 2020, 11, 4058. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.E.; Winney, I.S.; Salles, O.C.; Pujol, B. A guide to using a multiple-matrix animal model to disentangle genetic and nongenetic causes of phenotypic variance. PLoS ONE 2018, 13, e0197720. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Sork, V.L.; Cokus, S.J.; Fitz-Gibbon, S.T.; Zimin, A.V.; Puiu, D.; Garcia, J.A.; Gugger, P.F.; Henriquez, C.L.; Zhen, Y.; Lohmueller, K.E.; et al. High-quality genome and methylomes illustrate features underlying evolutionary success of oaks. bioRxiv 2021. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef]
- Pavlik, B.M.; Muick, P.C.; Johnson, S.G.; Popper, M. Oaks of California; Cachuma Press: Los Olivos, CA, USA, 1991. [Google Scholar]
- Anderson, M.K. Indigenous Uses, Management, and Restoration of Oaks of the Far Western United States; Natural Resources Conservation Service, National Plant Center, United States Department of Agriculture: Washington, DC, USA, 2007. [Google Scholar]
- Gugger, P.F.; Ikegami, M.; Sork, V.L. Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Nee. Mol. Ecol. 2013, 22, 3598–3612. [Google Scholar] [CrossRef]
- Sork, V.L.; Davis, F.W.; Westfall, R.; Flint, A.; Ikegami, M.; Wang, H.; Grivet, D. Gene movement and genetic association with regional climate gradients in California valley oak (Quercus lobata Nee) in the face of climate change. Mol. Ecol. 2010, 19, 3806–3823. [Google Scholar] [CrossRef]
- Delfino Mix, A.; Wright, J.W.; Gugger, P.F.; Liang, C.; Sork, V.L. Establishing a range-wide provenance test in valley oak (Quercus lobata Née) at two California sites. In Proceedings of the Seventh Oak Symposium: Managing Oak Woodlands in a Dynamic World; General Technical Report PSW-GTR-251; Standiford, R.B., Purcelll, K.L., Eds.; U.S. Department of Agriculture, Forest Service, Pacific Southwest Research Station: Berkeley, CA, USA, 2015; pp. 413–424. [Google Scholar]
- MacDonald, B.W. Local Climatic Heterogeneity Predicts Differences in Phenotypic Plasticity across Populations of a Widely-Distributed California Oak Species. Master’s Thesis, University of California, Los Angeles, CA, USA, 2017. [Google Scholar]
- Sage, R.D.; Koenig, W.D.; McLaughlin, B.C. Fitness consequences of seed size in the valley oak Quercus lobata Née (Fagaceae). Ann. For. Sci. 2011, 68, 477. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Sisó, S.; Camarero, J.; Gil-Pelegrín, E. Relationship between hydraulic resistance and leaf morphology in broadleaf Quercus species: A new interpretation of leaf lobation. Trees 2001, 15, 341–345. [Google Scholar] [CrossRef]
- Pérez-Harguindeguy, N.; Díaz, S.; Lavorel, S.; Poorter, H.; Jaureguiberry, P.; Bret-Harte, M.S.; Cornwell, W.K.; Craine, J.M.; Gurvich, D.E.; Urcelay, C.; et al. New handbook for standardized measurment of plant functional traits worldwide. Aust. J. Bot. 2013, 23, 167–234. [Google Scholar] [CrossRef]
- Niinemets, Ü. Global-scale climatic controls of leaf dry mass per area, density, and thickness in trees and shrubs. Ecology 2001, 82, 453–469. [Google Scholar] [CrossRef]
- Gianfagna, T.J.; Carter, C.D.; Sacalis, J.N. Temperature and photoperiod influence trichome density and sesquiterpene content of Lycopersicon hirsutum f. Hirsutum. Plant Physiol. 1992, 100, 1403–1405. [Google Scholar] [CrossRef]
- Picotte, J.J.; Rosenthal, D.M.; Rhode, J.M.; Cruzan, M.B. Plastic responses to temporal variation in moisture availability: Consequences for water use efficiency and plant performance. Oecologia 2007, 153, 821–832. [Google Scholar] [CrossRef]
- Ning, P.; Wang, J.; Zhou, Y.; Gao, L.; Wang, J.; Gong, C. Adaptional evolution of trichome in Caragana korshinskii to natural drought stress on the Loess Plateau, China. Ecol. Evol. 2016, 6, 3786–3795. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.M.; Walker, S.C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Li, J.T.; Yang, J.; Chen, D.C.; Zhang, X.L.; Tang, Z.S. An optimized mini-preparation method to obtain high-quality genomic DNA from mature leaves of sunflower. Genet. Mol. Res. 2007, 6, 1064–1071. [Google Scholar]
- Feng, S.; Rubbi, L.; Jacobsen, S.E.; Pellegrini, M. Determining DNA methylation profiles using sequencing. Methods Mol. Biol. 2011, 733, 223–238. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Eyring, K.; De, S.; Yang, I.V.; Schwartz, D.A. Fast and accurate alignment of long bisulfite-seq reads. arXiv 2014, arXiv:1401.1129. [Google Scholar]
- Sork, V.L.; Cokus, S.J.; Fitz-Gibbon, S.T.; Zimin, A.; Puiu, D.; Pellegrini, M.; Salzberg, S.L. Valley Oak Genomic Resources. Available online: https://valleyoak.ucla.edu/genomic-resources/ (accessed on 31 October 2019).
- Andri, S. DescTools: Tools for Descriptive Statistics. Available online: https://cran.r-project.org/web/packages/DescTools/index.html (accessed on 31 October 2019).
- Lea, A.J.; Vilgalys, T.P.; Durst, P.A.P.; Tung, J. Maximizing ecological and evolutionary insight in bisulfite sequencing data sets. Nat. Ecol. Evol. 2017, 1, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A Map Reduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Chen, W.; Zhu, Z.; Zhang, Q.; Nabais, M.F.; Qi, T.; Deary, I.J.; Wray, N.R.; Visscher, P.M.; McRae, A.F.; et al. OSCA: A tool for omic-data-based complex trait analysis. Genome Biol. 2019, 20, 107. [Google Scholar] [CrossRef]
- Main, B.J.; Lee, Y.; Ferguson, H.M.; Kreppel, K.S.; Kihonda, A.; Govella, N.J.; Collier, T.C.; Cornel, A.J.; Eskin, E.; Kang, E.Y.; et al. The genetic basis of host preference and resting behavior in the major African malaria vector, Anopheles arabiensis. PLoS Genet. 2016, 12, e1006303. [Google Scholar] [CrossRef]
- Browne, L.; Wright, J.W.; Fitz-Gibbon, S.; Gugger, P.F.; Sork, V.L. Adaptational lag to temperature in valley oak (Quercus lobata) can be mitigated by genome-informed assisted gene flow. Proc. Natl. Acad. Sci. USA 2019, 116, 25179–25185. [Google Scholar] [CrossRef]
- Bossdorf, O.; Arcuri, D.; Richards, C.L.; Pigliucci, M. Experimental alteration of DNA methylation affects the phenotypic plasticity of ecologically relevant traits in Arabidopsis thaliana. Evol. Ecol. 2010, 24, 541–553. [Google Scholar] [CrossRef]
- Kanchanaketu, T.; Hongtrakul, V. Treatment of 5-azacytidine as DNA demethylating agent in Jatropha curcas L. Kasetsart J-Nat. Sci. 2015, 49, 524–535. [Google Scholar]
- MacDonald, B.W.S.; Wright, J.W.; Sork, V.L. Local climatic Heterogeneity Predicts Differences in Phenotypic Plasticity across Populations of a Widely-Distributed California Oak, Quercus lobata Nee; In preparation.
- Ramirez, H.-L.; Ivey, C.T.; Wright, J.W.; MacDonald, B.W.S.; Sork, V.L. Variation in leaf shape in a Quercus lobata common garden: Tests for adaptation to climate and physiological consequences. Madrono 2020, 67, 77–84. [Google Scholar] [CrossRef]
- Griffin, P.T.; Niederhuth, C.E.; Schmitz, R.J. A comparative analysis of 5-Azacytidine- and zebularine-induced DNA demethylation. G3 2016, 6, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hemani, G.; Vinkhuyzen, A.A.E.; Chen, G.-B.; Lee, S.H.; Wray, N.R.; Goddard, M.E.; Yang, J. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 2014, 10, e1004269. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Yang, J.; Hayes, B.J.; Price, A.L.; Goddard, M.E.; Visscher, P.M. Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 2013, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trait | Height | Leaf Area | Leaf Lobedness | Specific Leaf Area | Leaf Thickness | Trichome Density | Powdery Mildew |
|---|---|---|---|---|---|---|---|
| Height | 1.00 | ||||||
| Leaf area | 0.46 | 1.00 | |||||
| Leaf lobedness | 0.25 | 0.34 | 1.00 | ||||
| Specific leaf area | −0.14 | 0.04 | 0.01 | 1.00 | |||
| Leaf thickness | −0.15 | 0.18 | −0.10 | −0.19 | 1.00 | ||
| Trichome density | 0.29 | −0.06 | −0.19 | −0.19 | −0.05 | 1.00 | |
| Powdery mildew | 0.44 | 0.17 | −0.05 | −0.06 | −0.11 | 0.16 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Browne, L.; MacDonald, B.; Fitz-Gibbon, S.; Wright, J.W.; Sork, V.L. Genome-Wide Variation in DNA Methylation Predicts Variation in Leaf Traits in an Ecosystem-Foundational Oak Species. Forests 2021, 12, 569. https://doi.org/10.3390/f12050569
Browne L, MacDonald B, Fitz-Gibbon S, Wright JW, Sork VL. Genome-Wide Variation in DNA Methylation Predicts Variation in Leaf Traits in an Ecosystem-Foundational Oak Species. Forests. 2021; 12(5):569. https://doi.org/10.3390/f12050569
Chicago/Turabian StyleBrowne, Luke, Brandon MacDonald, Sorel Fitz-Gibbon, Jessica W. Wright, and Victoria L. Sork. 2021. "Genome-Wide Variation in DNA Methylation Predicts Variation in Leaf Traits in an Ecosystem-Foundational Oak Species" Forests 12, no. 5: 569. https://doi.org/10.3390/f12050569
APA StyleBrowne, L., MacDonald, B., Fitz-Gibbon, S., Wright, J. W., & Sork, V. L. (2021). Genome-Wide Variation in DNA Methylation Predicts Variation in Leaf Traits in an Ecosystem-Foundational Oak Species. Forests, 12(5), 569. https://doi.org/10.3390/f12050569