Molecular Signatures of Reticulate Evolution within the Complex of European Pine Taxa


Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Genotyping
2.2. Genetic Diversity and Structure
2.3. Phylogenetic Analyses
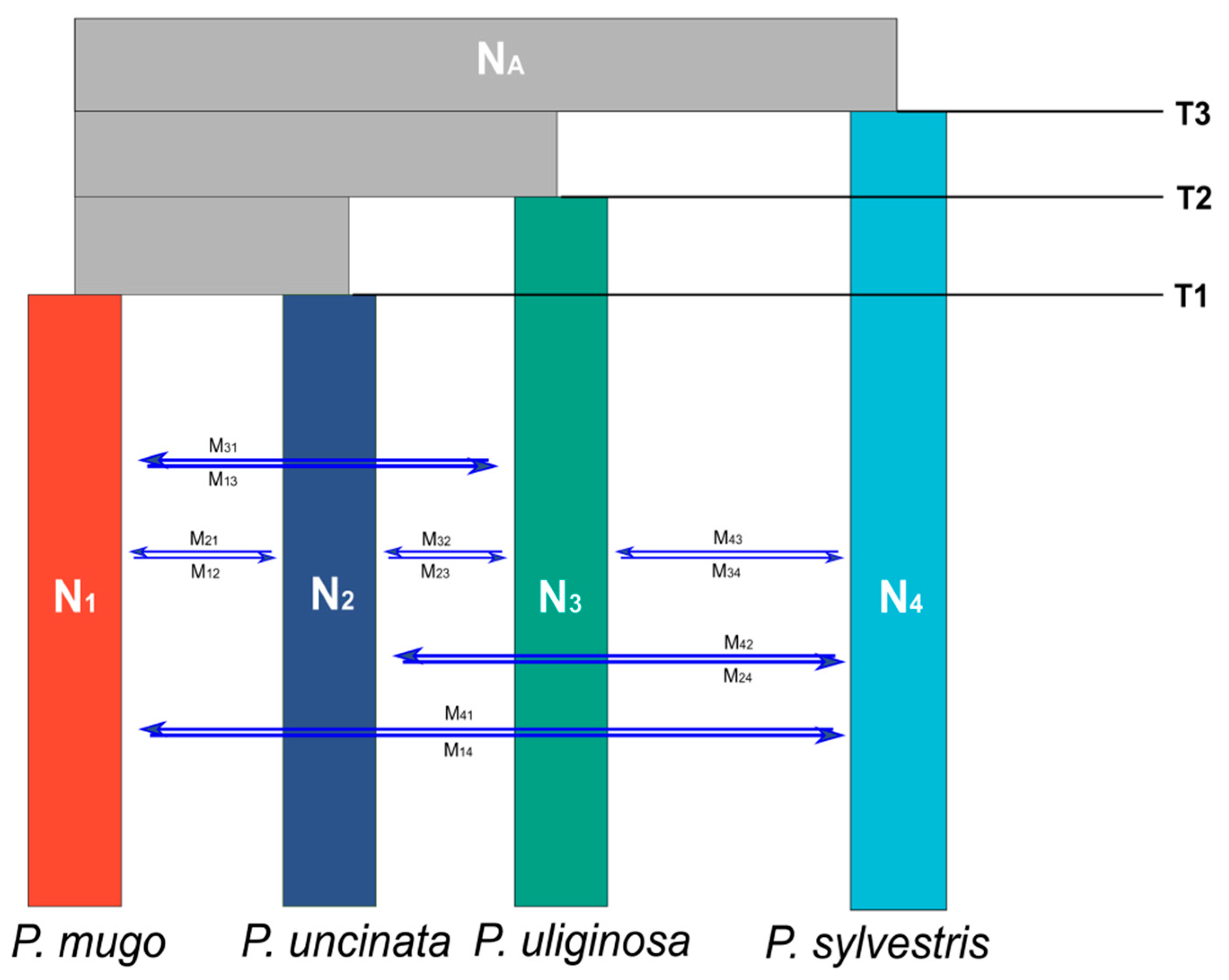
2.4. Testing Speciation Models Using Coalescent Simulations
3. Results
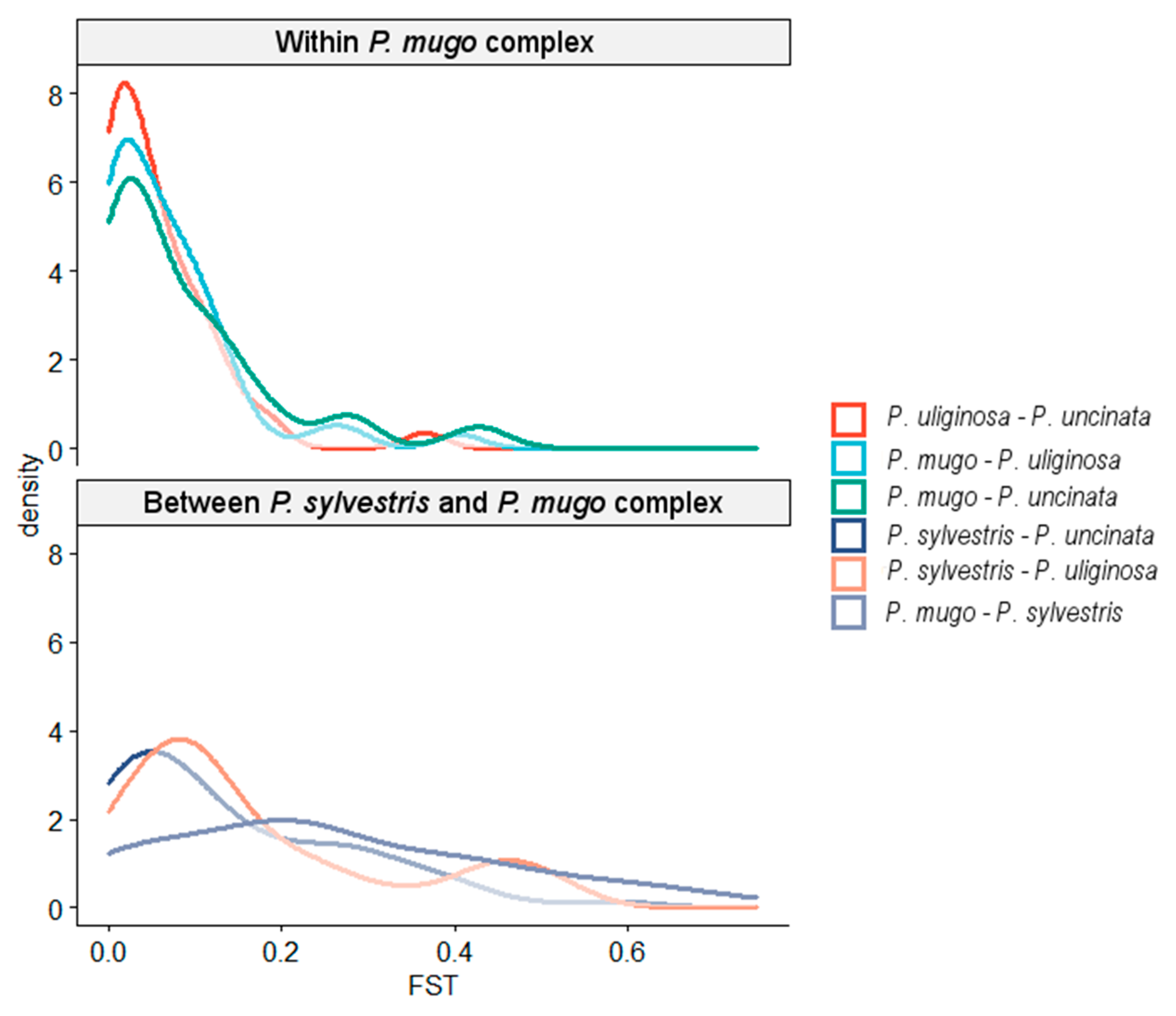
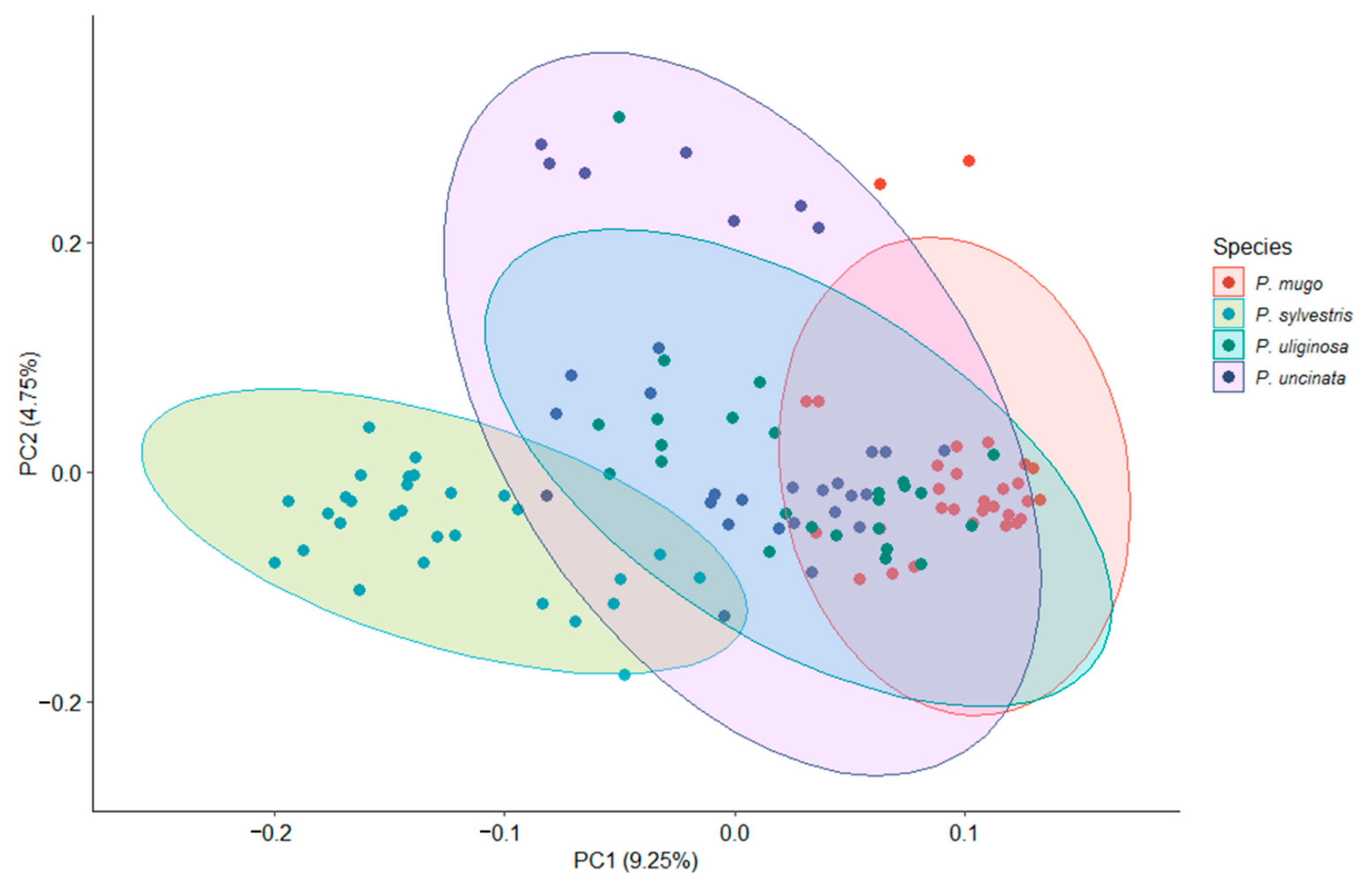
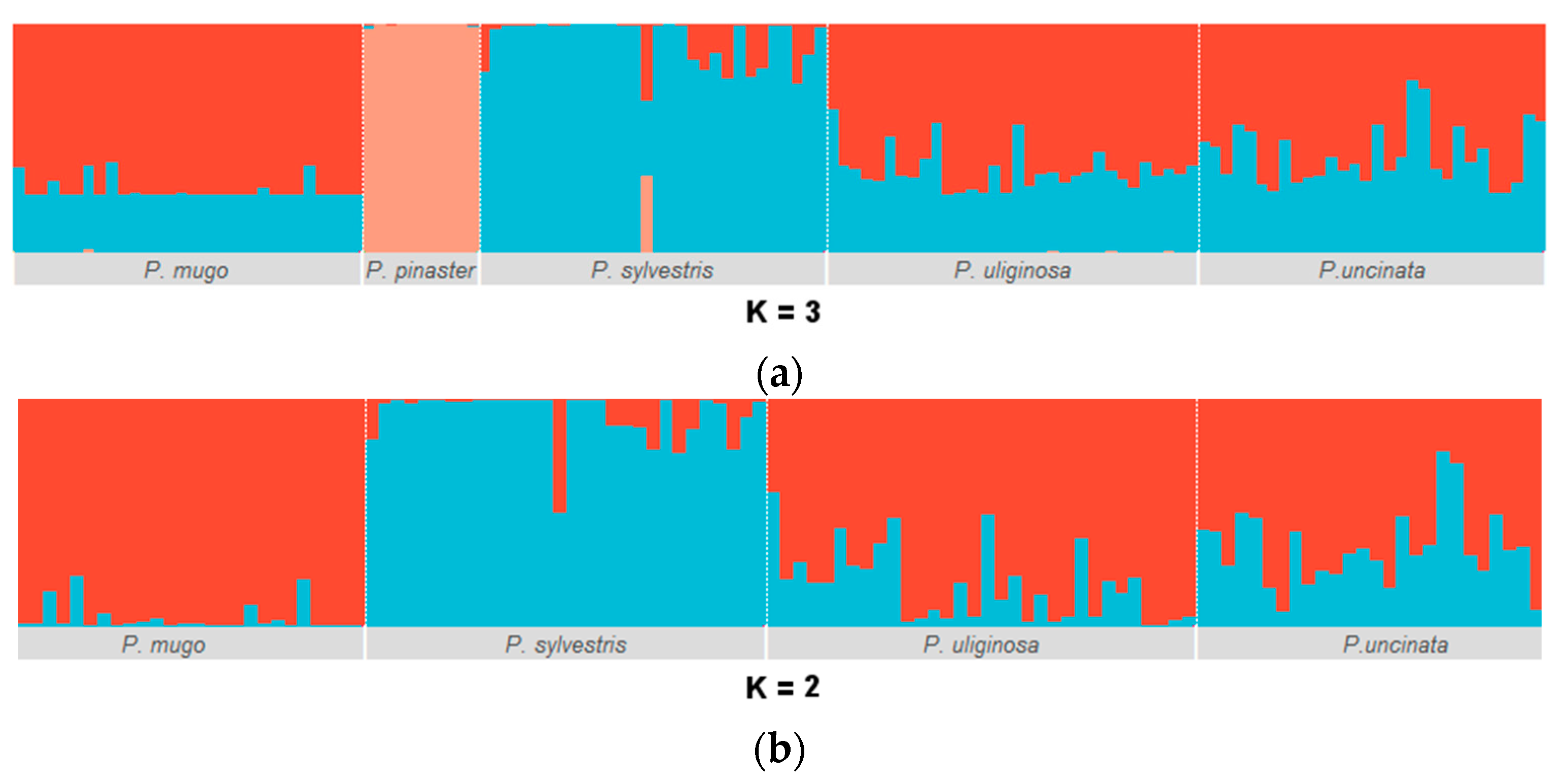
3.1. Genetic Diversity and Population Structure
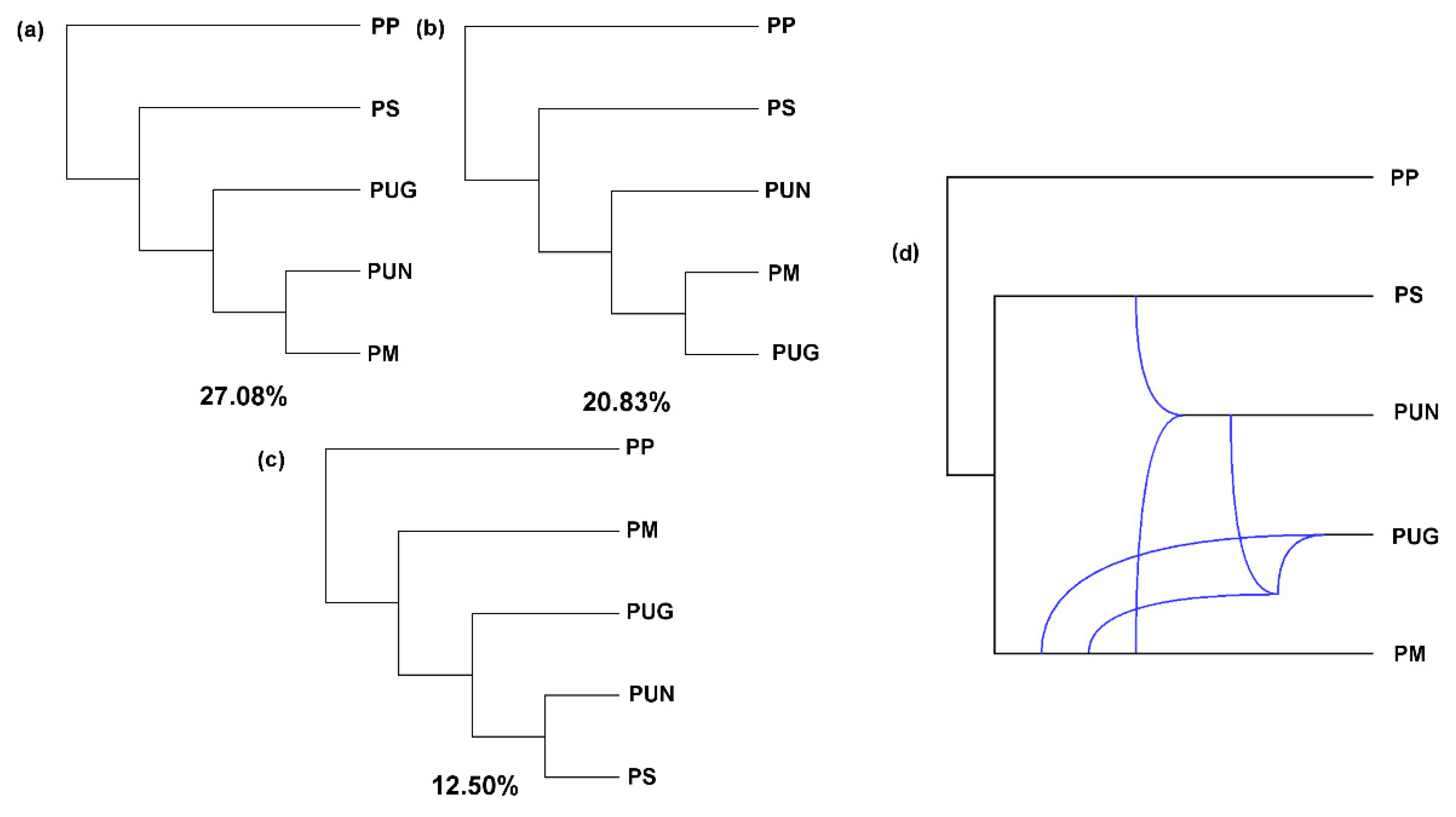
3.2. Reticulate Phylogeny of Four Pines
3.3. Alternative Speciation Models
4. Discussion
4.1. Models of Speciation
4.2. Interspecific Gene Flow
4.3. Species Integrity within the P. mugo Complex
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hendry, A.P. Ecological speciation! Or the lack thereof? Can. J. Fish. Aquat. Sci. 2009, 66, 1383–1398. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization, ecological races and the nature of species: Empirical evidence for the ease of speciation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 2971–2986. [Google Scholar] [CrossRef]
- Galtier, N. Delineating species in the speciation continuum: A proposal. Evol. Appl. 2019, 12, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Nosil, P. Speciation with gene flow could be common. Mol. Ecol. 2008, 17, 2103–2106. [Google Scholar] [CrossRef]
- Feder, J.L.; Egan, S.P.; Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 2012, 28, 342–350. [Google Scholar] [CrossRef]
- Martin, S.H.; Dasmahapatra, K.K.; Nadeau, N.J.; Salazar, C.; Walters, J.R.; Simpson, F.; Blaxter, M.; Manica, A.; Mallet, J.; Jiggins, C.D. Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 2013, 23, 1817–1828. [Google Scholar] [CrossRef]
- Gailing, O.; Curtu, A.L. Interspecific gene flow and maintenance of species integrity in oaks. Ann. For. Res. 2014, 57, 5–18. [Google Scholar] [CrossRef]
- Schumer, M.; Rosenthal, G.G.; Andolfatto, P. How common is homoploid hybrid speciation? Evolution 2014, 68, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, B.S.; Mao, I.F.; Ingvarsson, P.; Zeng, Q.Y.; Wang, X.R. Demography and speciation history of the homoploid hybrid pine Pinus densata on the Tibetan Plateau. Mol. Ecol. 2012, 21, 4811–4827. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.L. Evolution through Genetic Exchange; Oxford University Press: Oxford, UK, 2007; pp. 1–252. [Google Scholar] [CrossRef]
- Lamichhaney, S.; Han, F.; Webster, M.T.; Andersson, L.; Grant, B.R.; Grant, P.R. Rapid hybrid speciation in Darwin’s finches. Science 2018, 359, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Ru, D.; Sun, Y.; Wang, D.; Chen, Y.; Wang, T.; Hu, Q.; Abbott, R.J.; Liu, J. Population genomic analysis reveals that homoploid hybrid speciation can be a lengthy process. Mol. Ecol. 2018, 27, 4875–4887. [Google Scholar] [CrossRef]
- Mallet, J. Hybridization as an invasion of the genome. Trends Ecol. Evol. 2005, 20, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Nieto Feliner, G.; Álvarez, I.; Fuertes-Aguilar, J.; Heuertz, M.; Marques, I.; Moharrek, F.; Piñeiro, R.; Riina, R.; Rosselló, J.A.; Soltis, P.S.; et al. Is homoploid hybrid speciation that rare? An empiricist’s view. Heredity 2017, 118, 513–516. [Google Scholar] [CrossRef]
- Harrison, R.G.; Larson, E.L. Hybridization, introgression, and the nature of species boundaries. J. Hered. 2014, 105, 795–809. [Google Scholar] [CrossRef]
- Nosil, P.; Funk, D.J.; Ortiz-Barrientos, D. Divergent selection and heterogeneous genomic divergence. Mol. Ecol. 2009, 18, 375–402. [Google Scholar] [CrossRef]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef]
- Grant, P.R.; Grant, B.R. Unpredictable Evolution in a 30-Year Study of Darwin’s Finches. Science 2002, 296, 707–711. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Ward, T.J.; Robert, V.A.R.G.; Crous, P.W.; Geiser, D.M.; Kang, S. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica 2015, 43, 583–595. [Google Scholar] [CrossRef]
- Harbach, R.E. Culex pipiens: Species versus species complex taxonomic history and perspective. J. Am. Mosq. Control Assoc. 2012, 28, 10–23. [Google Scholar] [CrossRef]
- Welch, M.E.; Rieseberg, L.H. Habitat divergence between a homoploid hybrid sunflower species, Helianthus paradoxus (Asteraceae), and its progenitors. Am. J. Bot. 2002, 89, 472–478. [Google Scholar] [CrossRef]
- Crespo, M.; Azorín, M.; Mavrodiev, E. Can a rainbow consist of a single colour? A new comprehensive generic arrangement of the ‘Iris sensu latissimo‘ clade (Iridaceae), congruent with morphology and molecular data. Phytotaxa 2015, 232, 1–78. [Google Scholar] [CrossRef]
- Bucci, G.; Anzidei, M.; Madaghiele, A.; Vendramin, G.G. Detection of haplotypic variation and natural hybridization in halepensis-complex pine species using chloroplast simple sequence repeat (SSR) markers. Mol. Ecol. 1998, 7, 1633–1643. [Google Scholar] [CrossRef]
- Yeh, F.; Ye, T.; Yang, R.-C. Multilocus structure in the Pinus contorta–Pinus banksiana complex. Can. J. Bot. 2007, 85, 774–784. [Google Scholar] [CrossRef]
- Sun, Y.; Abbott, R.J.; Lu, Z.; Mao, K.; Zhang, L.; Wang, X.; Ru, D.; Liu, J. Reticulate evolution within a spruce (Picea) species complex revealed by population genomic analysis. Evolution 2018, 72, 2669–2681. [Google Scholar] [CrossRef]
- Moeller, M.; Gao, L.-M.; Mill, R.; Liu, J.; Zhang, D.-Q.; Poudel, R.; Li, D.-Z. A multidisciplinary approach reveals hidden taxonomic diversity in the morphologically challenging Taxus wallichiana complex. Taxon 2013, 62, 1161–1177. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Knowles, L.L.; Carstens, B.C. Delimiting Species without Monophyletic Gene Trees. Syst. Biol. 2007, 56, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Naciri, Y.; Linder, H.P. Species delimitation and relationships: The dance of the seven veils. Taxon 2015, 64, 3–16. [Google Scholar] [CrossRef]
- Via, S. Natural selection in action during speciation. Proc. Natl. Acad. Sci. USA 2009, 106, 9939–9946. [Google Scholar] [CrossRef]
- Christensen, K.L. Taxonomic revision of the Pinus mugo complex and P. rhaetica (P. mugo sylvestris) (Pinaceae). Nord. J. Bot. 1987, 7, 383–408. [Google Scholar] [CrossRef]
- Farjon, A. A Handbook of the World’s Conifers (2 Vols.): Revised and Updated Edition; Brill: Leiden, The Netherlands, 2010. [Google Scholar]
- Łabiszak, B.; Zaborowska, J.; Wachowiak, W. Patterns of mtDNA variation reveal complex evolutionary history of relict and endangered peat bog pine (Pinus uliginosa). AoB Plants 2019, 11. [Google Scholar] [CrossRef]
- Wachowiak, W.; Zukowska, W.B.; Wojkiewicz, B.; Cavers, S.; Litkowiec, M. Hybridization in contact zone between temperate European pine species. Tree Genet. Genomes 2016, 12. [Google Scholar] [CrossRef]
- Businský, R.; Kirschner, J. Pinus mugo and P. uncinata as parents of hybrids a taxonomic and nomenclatural survey. Phyton 2010, 50, 27–57. [Google Scholar]
- Wachowiak, W.; Palme, A.E.; Savolainen, O. Speciation history of three closely related pines Pinus mugo (T.), P. uliginosa (N.) and P. sylvestris (L.). Mol. Ecol. 2011, 20, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Dzialuk, A.; Boratyńska, K.; Romo, A.; Boratyński, A. Taxonomic and geographic variation of the Pinus mugo complex on chloroplast microsatellite markers. Syst. Biodivers. 2017, 15, 464–479. [Google Scholar] [CrossRef]
- Krzakowa, M.; Naganowska, B.; Bobowicz, M.A. Investigations on taxonomic status of Pinus uliginosa Neumann. Bull. Soc. Pol. Acad. Sci. 1984, 24, 87–96. [Google Scholar]
- Boratynska, K.; Boratynski, A. Taxonomic differences among closely related pines Pinus sylvestris, P. mugo, P. uncinata, P. rotundata and P. uliginosa as revealed in needle sclerenchyma cells. Flora 2007, 202, 555–569. [Google Scholar] [CrossRef]
- Lewandowski, A.; Boratynski, A.; Mejnartowicz, L. Allozyme investigations on the genetic differentiation between closely related pines—Pinus sylvestris, P. mugo, P. uncinata, and P. uliginosa (Pinaceae). Plant Syst. Evol. 2000, 221, 15–24. [Google Scholar] [CrossRef]
- Żukowska, W.B.; Wachowiak, W. Utility of closely related taxa for genetic studies of adaptive variation and speciation: Current state and perspectives in plants with focus on forest tree species. J. Syst. Evol. 2016, 54, 17–28. [Google Scholar] [CrossRef]
- Newton, A.C.; Allnutt, T.R.; Gillies, A.C.M.; Lowe, A.J.; Ennos, R.A. Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol. Evol. 1999, 14, 140–145. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics, 2nd ed.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2010; 618p. [Google Scholar]
- Allendorf, F.W.; Luikart, G.H.; Aitken, S.N. Conservation and the Genetics of Populations; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Wachowiak, W.; Zaborowska, J.; Labiszak, B.; Perry, A.; Zucca, G.M.; Gonzalez-Martinez, S.C.; Cavers, S. Molecular signatures of divergence and selection in closely related pine taxa. Tree Genet. Genomes 2018, 14, 83. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Hey, J.; Wakeley, J. A coalescent estimator of the population recombination rate. Genetics 1997, 145, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A. Ggpubr: ‘Ggplot2’ Based Publication Ready Plots. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 15 January 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org (accessed on 15 December 2020).
- Tang, Y.; Horikoshi, M.; Li, W.X. ggfortify: Unified Interface to Visualize Statistical Results of Popular R Packages. R J. 2016, 8, 474–485. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Than, C.; Ruths, D.; Nakhleh, L. PhyloNet: A software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinform. 2008, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Yu, Y.; Zhu, J.; Nakhleh, L. Inferring Phylogenetic Networks Using PhyloNet. Syst. Biol. 2018, 67, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Elworth, R.; Ogilvie, H.; Zhu, J.; Nakhleh, L. Advances in Computational Methods for Phylogenetic Networks in the Presence of Hybridization. Bioinform. Phylogenet. 2019, 29, 317–360. [Google Scholar] [CrossRef]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for Ancient Admixture between Closely Related Populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.J.; van Oosterhout, C. hybridcheck: Software for the rapid detection, visualization and dating of recombinant regions in genome sequence data. Mol. Ecol. Resour. 2016, 16, 534–539. [Google Scholar] [CrossRef]
- Excoffier, L.; Foll, M. fastsimcoal: A continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 2011, 27, 1332–1334. [Google Scholar] [CrossRef]
- Excoffier, L.; Dupanloup, I.; Huerta-Sanchez, E.; Sousa, V.C.; Foll, M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Meier, J. Fsc-selectbestrun.sh. GitHub Repository. 2018. Available online: https://github.com/speciationgenomics/scripts/blob/master/fsc-selectbestrun.sh (accessed on 15 December 2020).
- Meier, J. calculateAIC.R. GitHub Repository. 2018. Available online: https://github.com/speciationgenomics/scripts/blob/master/calculateAIC.sh (accessed on 15 December 2020).
- Li, Y.; Stocks, M.; Hemmila, S.; Kallman, T.; Zhu, H.; Zhou, Y.; Chen, J.; Liu, J.; Lascoux, M. Demographic histories of four spruce (Picea) species of the Qinghai-Tibetan Plateau and neighboring areas inferred from multiple nuclear loci. Mol. Biol. Evol. 2010, 27, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, A.R.; Li, Z.; Van de Peer, Y.; Ingvarsson, P.K. Contrasting rates of molecular evolution and patterns of selection among Gymnosperms and flowering plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.; Hinkley, D. Bootstrap Methods and Their Application. J. Am. Stat. Assoc. 1997, 94. [Google Scholar] [CrossRef]
- Rosenberg, N.A. The shapes of neutral gene genealogies in two species: Probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution 2003, 57, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Heuertz, M.; Teufel, J.; González-Martínez, S.C.; Soto, A.; Fady, B.; Alía, R.; Vendramin, G.G. Geography determines genetic relationships between species of mountain pine (Pinus mugo complex) in western Europe. J. Biogeogr. 2010, 37, 541–556. [Google Scholar] [CrossRef]
- Łabiszak, B.; Zaborowska, J.; Wójkiewicz, B.; Wachowiak, W. Molecular and paleo-climatic data uncover the impact of an ancient bottleneck on the demographic history and contemporary genetic structure of endangered Pinus uliginosa. J. Syst. Evol. 2019. [Google Scholar] [CrossRef]
- Willyard, A.; Cronn, R.; Liston, A. Reticulate evolution and incomplete lineage sorting among the Ponderosa pines. Mol. Phylogenet. Evol. 2009, 52, 498–511. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.I.; Ru, D.; Miao, J.; Tao, W.; Zhang, L.; Xu, J.; Liu, J.; Mao, K. Allopatric divergence and hybridization within Cupressus chengiana (Cupressaceae), a threatened conifer in the northern Hengduan Mountains of western China. Mol. Ecol. 2020, 29, 1250–1266. [Google Scholar] [CrossRef]
- Wachowiak, W.; Prus-Glowacki, W. Hybridisation processes in sympatric populations of pines Pinus sylvestris L., P. mugo Turra and P. uliginosa Neumann. Plant Syst. Evol. 2008, 271, 29–40. [Google Scholar] [CrossRef]
- Sobierajska, K.; Wachowiak, W.; Zaborowska, J.; Labiszak, B.; Wojkiewicz, B.; Sekiewicz, M.; Jasinska, A.K.; Sekiewicz, K.; Boratynska, K.; Marcysiak, K.; et al. Genetic consequences of hybridization in relict isolated trees Pinus sylvestris and the Pinus mugo complex. Forests 2020, 11, 1086. [Google Scholar] [CrossRef]
- Kormutak, A.; Galgoci, M.; Sukenikova, D.; Bolecek, P.; Libantova, J.; Gőmőry, D. Maternal inheritance of chloroplast DNA in Pinus mugo Turra: A case study of Pinus mugo × Pinus sylvestris crossing. Plant Syst. Evol. 2018, 304, 71–76. [Google Scholar] [CrossRef]
- Zaborowska, J.; Łabiszak, B.; Wachowiak, W. Population history of European mountain pines Pinus mugo and Pinus uncinata revealed by mitochondrial DNA markers. J. Syst. Evol. 2019. [Google Scholar] [CrossRef]
- McLachlan, J.S.; Clark, J.S. Reconstructing historical ranges with fossil data at continental scales. For. Ecol. Manag. 2004, 197, 139–147. [Google Scholar] [CrossRef]
- Willis, K.J.; van Andel, T.H. Trees or no trees? The Environments of central and eastern Europe during the last glaciation. Quat. Sci. Rev. 2004, 23. [Google Scholar] [CrossRef]
- Wojkiewicz, B.; Litkowiec, M.; Wachowiak, W. Contrasting patterns of genetic variation in core and peripheral populations of highly outcrossing and wind pollinated forest tree species. Aob Plants 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Hebda, A.; Wójkiewicz, B.; Wachowiak, W. Genetic characteristics of Scots pine in Poland and reference populations based on nuclear and chloroplast microsatellite markers. Silva Fenn. 2017, 51. [Google Scholar] [CrossRef]
- Baack, E.J.; Rieseberg, L.H. A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 2007, 17, 513–518. [Google Scholar] [CrossRef]
- Kwit, C.; Moon, H.S.; Warwick, S.I.; Stewart, C.N. Transgene introgression in crop relatives: Molecular evidence and mitigation strategies. Trends Biotechnol. 2011, 29, 284–293. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Meirmans, P.; Rong, J.; Bartsch, D.; Ghosh, A.; de Jong, T.J.; Haccou, P.; Lu, B.-R.; Snow, A.A.; Neal Stewart, C.; et al. Introgression of crop alleles into wild or weedy populations. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 325–345. [Google Scholar] [CrossRef]
- Snow, A.A.; Pilson, D.; Rieseberg, L.H.; Paulsen, M.J.; Pleskac, N.; Reagon, M.R.; Wolf, D.E.; Selbo, S.M. A Bt transgene reduces herbivory and enhances fecundity in wild sunflowers. Ecol. Appl. 2003, 13, 279–286. [Google Scholar] [CrossRef]
- Hufford, M.B.; Lubinksy, P.; Pyhäjärvi, T.; Devengenzo, M.T.; Ellstrand, N.C.; Ross-Ibarra, J. The genomic signature of crop-wild introgression in maize. PLoS Genet. 2013, 9, e1003477. [Google Scholar] [CrossRef]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? Bioessays 2016, 38, 140–149. [Google Scholar] [CrossRef]
- Wachowiak, W.; Perry, A.; Donnelly, K.; Cavers, S. Early phenology and growth trait variation in closely related European pine species. Ecol. Evol. 2018, 8, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Wachowiak, W.; Brown, A.V.; Ennos, R.A.; Cottrell, J.E.; Cavers, S. Substantial heritable variation for susceptibility to Dothistroma septosporum within populations of native British Scots pine (Pinus sylvestris). Plant Pathol. 2016, 65, 987–996. [Google Scholar] [CrossRef]
- Ortiz-Barrientos, D.; Baack, E.J. Species integrity in trees. Mol. Ecol. 2014, 23, 4188–4191. [Google Scholar] [CrossRef] [PubMed]
- Harr, B. Genomic islands of differentiation between house mouse subspecies. Genome Res. 2006, 16, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Grassa, C.J.; Yeaman, S.; Moyers, B.T.; Lai, Z.; Kane, N.C.; Bowers, J.E.; Burke, J.M.; Rieseberg, L.H. Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nat. Commun. 2013, 4, 1827. [Google Scholar] [CrossRef]
- Michel, A.P.; Sim, S.; Powell, T.H.; Taylor, M.S.; Nosil, P.; Feder, J.L. Widespread genomic divergence during sympatric speciation. Proc. Natl. Acad. Sci. USA 2010, 107, 9724–9729. [Google Scholar] [CrossRef] [PubMed]
- Poelstra, J.W.; Vijay, N.; Bossu, C.M.; Lantz, H.; Ryll, B.; Muller, I.; Baglione, V.; Unneberg, P.; Wikelski, M.; Grabherr, M.G.; et al. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 2014, 344, 1410–1414. [Google Scholar] [CrossRef] [PubMed]
- Wachowiak, W.; Boratynska, K.; Cavers, S. Geographical patterns of nucleotide diversity and population differentiation in three closely related European pine species in the Pinus mugo complex. Bot. J. Linn. Soc. 2013, 172, 225–238. [Google Scholar] [CrossRef][Green Version]
- Neale, D.B.; Wegrzyn, J.L.; Stevens, K.A.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; Holtz-Morris, A.E.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Wachowiak, W.; Downing, A.; Talbot, R.; Cavers, S. Development of a single nucleotide polymorphism array for population genomic studies in four European pine species. Mol. Ecol. Resour. 2020, 20, 1697–1705. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | n | L (bp) | P | S | π | D | R | Ks | Hd (SD) |
|---|---|---|---|---|---|---|---|---|---|
| P. uliginosa | 29.3 | 19,414 | 441 | 155 | 0.005245 | −0.28 | 39 | 1.864 | 0.689 (0.066) |
| P. mugo | 29.3 | 19,414 | 365 | 154 | 0.004039 | −0.37 | 8 | 1.501 | 0.601 (0.071) |
| P. uncinata | 29.6 | 19,414 | 363 | 113 | 0.004546 | −0.14 | 8 | 1.656 | 0.623 (0.066) |
| P. sylvestris | 27.8 | 19,414 | 367 | 128 | 0.004576 | −0.16 | 14 | 1.682 | 0.598 (0.073) |
| Species Pair | FST ± SD | Dnet ± SD |
|---|---|---|
| P. uliginosa vs. P. mugo | 0.068 ± 0.088 | 0.00032 ± 0.00042 |
| P. uliginosa vs. P. uncinata | 0.056 ± 0.067 | 0.00032 ± 0.00046 |
| P. uliginosa vs. P. sylvestris | 0.167 ± 0.155 | 0.00108 ± 0.00109 |
| P. mugo vs. P. uncinata | 0.088 ± 0.104 | 0.00054 ± 0.00087 |
| P. sylvestris vs. P. mugo | 0.260 ± 0.197 | 0.00181 ± 0.00203 |
| P. sylvestris vs. P. uncinata | 0.142 ± 0.138 | 0.00084 ± 0.00110 |
| P1 | P2 | P3 | A | D | p | Z |
|---|---|---|---|---|---|---|
| PM | PUG | PUN | PP | 0.601 | ns | 1.073 |
| PM | PUN | PUG | PP | 0.161 | ns | 1.172 |
| PUN | PUG | PM | PP | 0.487 | <0.0001 | 3.304 |
| PM | PS | PUN | PP | −0.035 | ns | −0.128 |
| PUN | PS | PM | PP | 0.288 | <0.05 | 1.990 |
| PUN | PM | PS | PP | 0.320 | ns | 1.67 |
| PM | PS | PUG | PP | 0.516 | <0.0001 | 4.521 |
| PM | PUG | PS | PP | 0.641 | <0.0001 | 5.057 |
| PM | PUG | PUN | PS | 0.603 | ns | 1.073 |
| PM | PUN | PUG | PS | 0.183 | ns | 1.172 |
| PUN | PUG | PM | PS | 0.472 | <0.0001 | 3.304 |
| Parameters | Point Estimate | Lower Boundof 95% CI | Upper Boundof 95% CI |
|---|---|---|---|
| NA | 1,817,521 | 1,223,654 | 1,511,901 |
| N1 | 406,282 | 353,176 | 674,610 |
| N2 | 78,879 | 4839 | 88,690 |
| N3 | 77,680 | 5842 | 81,793 |
| N4 | 554,232 | 508,589 | 761,921 |
| T1 | 2,074,675 | 1,403,763 | 2,422,625 |
| T2 | 3,999,925 | 3,915,625 | 5,303,234 |
| T3 | 5,925,175 | 5,038,781 | 8,507,750 |
| M1→2 | 3.28−10 | 6.97−11 | 9.83−9 |
| M2→1 | 1.26−9 | 2.30−10 | 7.83−8 |
| M1→3 | 8.11−8 | 3.78−9 | 4.00−7 |
| M3→1 | 4.91−5 | 8.56−6 | 1.41−4 |
| M2→3 | 1.24−7 | 3.15−8 | 2.00−7 |
| M3→2 | 3.36−5 | 6.25−9 | 8.75−4 |
| M1→4 | 1.62−6 | 1.50−6 | 1.76−6 |
| M4→1 | 1.55−7 | 5.71−9 | 5.39−6 |
| M2→4 | 2.94−5 | 6.90−6 | 1.06−5 |
| M4→2 | 1.55−7 | 5.71−9 | 5.39−6 |
| M3→4 | 6.03−6 | 5.38−6 | 9.47−6 |
| M4→3 | 1.55−7 | 5.71−9 | 5.39−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łabiszak, B.; Wachowiak, W. Molecular Signatures of Reticulate Evolution within the Complex of European Pine Taxa. Forests 2021, 12, 489. https://doi.org/10.3390/f12040489
Łabiszak B, Wachowiak W. Molecular Signatures of Reticulate Evolution within the Complex of European Pine Taxa. Forests. 2021; 12(4):489. https://doi.org/10.3390/f12040489
Chicago/Turabian StyleŁabiszak, Bartosz, and Witold Wachowiak. 2021. "Molecular Signatures of Reticulate Evolution within the Complex of European Pine Taxa" Forests 12, no. 4: 489. https://doi.org/10.3390/f12040489
APA StyleŁabiszak, B., & Wachowiak, W. (2021). Molecular Signatures of Reticulate Evolution within the Complex of European Pine Taxa. Forests, 12(4), 489. https://doi.org/10.3390/f12040489